

Defining an essence of structure determining residue contacts in proteins
- PMID: 19997489
- PMCID: PMC2778133
- DOI: 10.1371/journal.pcbi.1000584
Defining an essence of structure determining residue contacts in proteins
Abstract
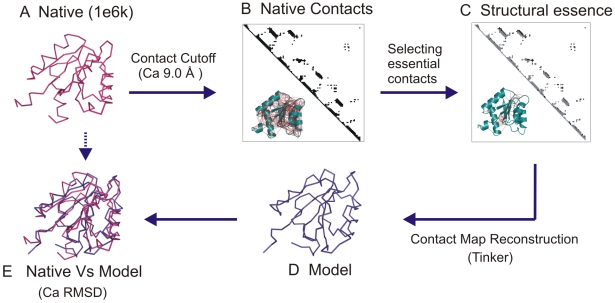
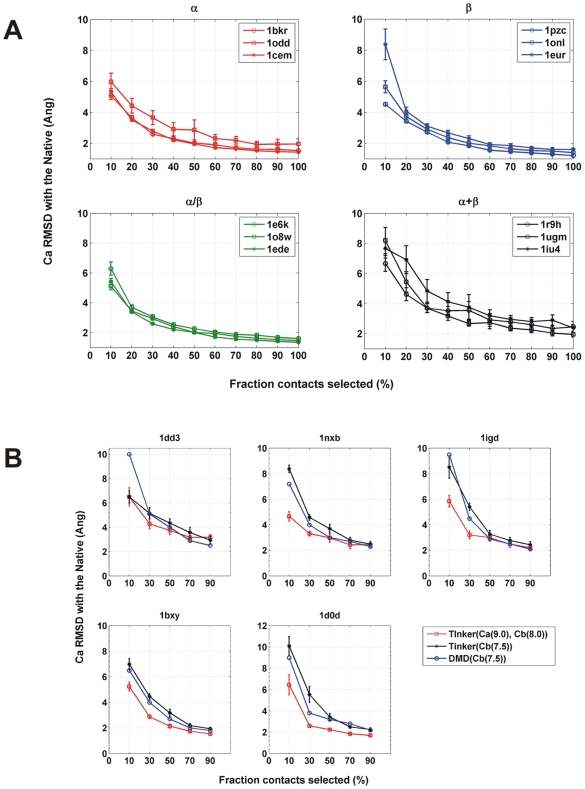
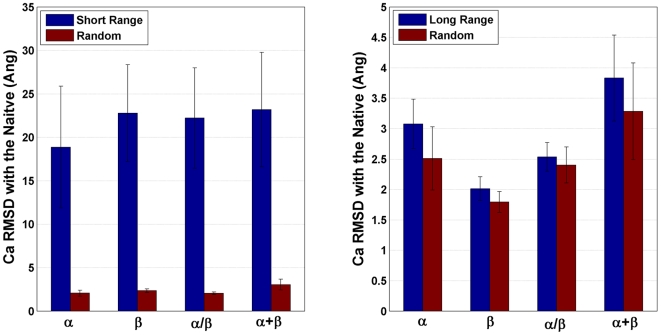
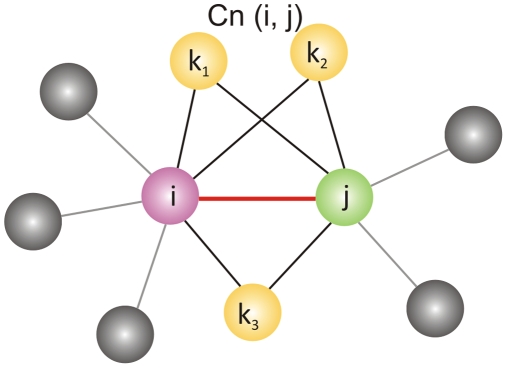
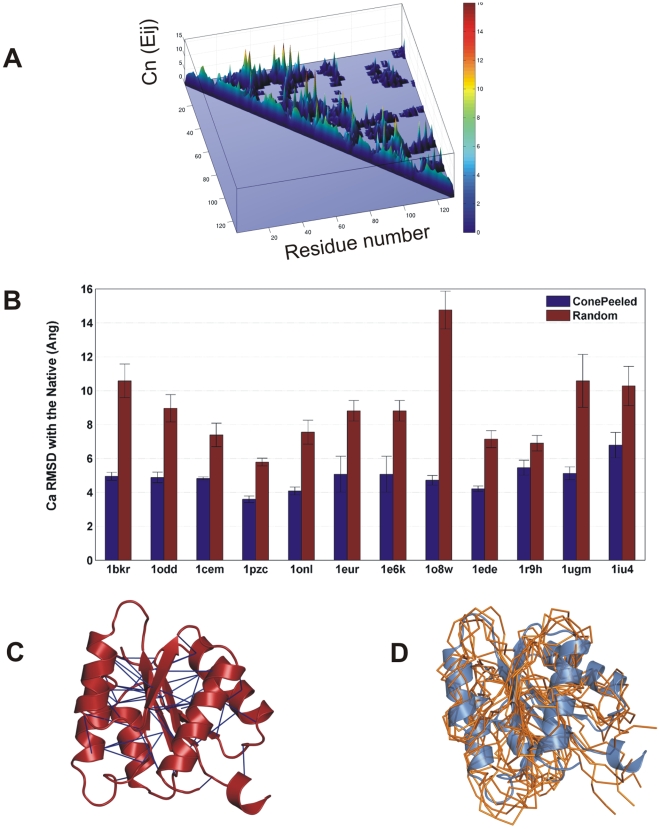
The network of native non-covalent residue contacts determines the three-dimensional structure of a protein. However, not all contacts are of equal structural significance, and little knowledge exists about a minimal, yet sufficient, subset required to define the global features of a protein. Characterisation of this "structural essence" has remained elusive so far: no algorithmic strategy has been devised to-date that could outperform a random selection in terms of 3D reconstruction accuracy (measured as the Ca RMSD). It is not only of theoretical interest (i.e., for design of advanced statistical potentials) to identify the number and nature of essential native contacts-such a subset of spatial constraints is very useful in a number of novel experimental methods (like EPR) which rely heavily on constraint-based protein modelling. To derive accurate three-dimensional models from distance constraints, we implemented a reconstruction pipeline using distance geometry. We selected a test-set of 12 protein structures from the four major SCOP fold classes and performed our reconstruction analysis. As a reference set, series of random subsets (ranging from 10% to 90% of native contacts) are generated for each protein, and the reconstruction accuracy is computed for each subset. We have developed a rational strategy, termed "cone-peeling" that combines sequence features and network descriptors to select minimal subsets that outperform the reference sets. We present, for the first time, a rational strategy to derive a structural essence of residue contacts and provide an estimate of the size of this minimal subset. Our algorithm computes sparse subsets capable of determining the tertiary structure at approximately 4.8 A Ca RMSD with as little as 8% of the native contacts (Ca-Ca and Cb-Cb). At the same time, a randomly chosen subset of native contacts needs about twice as many contacts to reach the same level of accuracy. This "structural essence" opens new avenues in the fields of structure prediction, empirical potentials and docking.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures

Similar articles
-
Toward an accurate prediction of inter-residue distances in proteins using 2D recursive neural networks.BMC Bioinformatics. 2014 Jan 10;15:6. doi: 10.1186/1471-2105-15-6. BMC Bioinformatics. 2014. PMID: 24410833 Free PMC article.
-
How well can we predict native contacts in proteins based on decoy structures and their energies?Proteins. 2003 Sep 1;52(4):598-608. doi: 10.1002/prot.10444. Proteins. 2003. PMID: 12910459
-
Fidelity of the protein structure reconstruction from inter-residue proximity constraints.J Phys Chem B. 2007 Jun 28;111(25):7432-8. doi: 10.1021/jp068963t. Epub 2007 Jun 2. J Phys Chem B. 2007. PMID: 17542631
-
Protein-protein docking tested in blind predictions: the CAPRI experiment.Mol Biosyst. 2010 Dec;6(12):2351-62. doi: 10.1039/c005060c. Epub 2010 Aug 19. Mol Biosyst. 2010. PMID: 20725658 Review.
-
TAPO: A combined method for the identification of tandem repeats in protein structures.FEBS Lett. 2015 Sep 14;589(19 Pt A):2611-9. doi: 10.1016/j.febslet.2015.08.025. Epub 2015 Aug 29. FEBS Lett. 2015. PMID: 26320412 Review.
Cited by
-
CONFOLD: Residue-residue contact-guided ab initio protein folding.Proteins. 2015 Aug;83(8):1436-49. doi: 10.1002/prot.24829. Epub 2015 Jun 6. Proteins. 2015. PMID: 25974172 Free PMC article.
-
Prediction of protein-binding areas by small-world residue networks and application to docking.BMC Bioinformatics. 2011 Sep 26;12:378. doi: 10.1186/1471-2105-12-378. BMC Bioinformatics. 2011. PMID: 21943333 Free PMC article.
-
Improved protein structure reconstruction using secondary structures, contacts at higher distance thresholds, and non-contacts.BMC Bioinformatics. 2017 Aug 29;18(1):380. doi: 10.1186/s12859-017-1807-5. BMC Bioinformatics. 2017. PMID: 28851269 Free PMC article.
-
Assessing Predicted Contacts for Building Protein Three-Dimensional Models.Methods Mol Biol. 2017;1484:115-126. doi: 10.1007/978-1-4939-6406-2_9. Methods Mol Biol. 2017. PMID: 27787823 Free PMC article.
-
Probabilistic grammatical model for helix-helix contact site classification.Algorithms Mol Biol. 2013 Dec 18;8(1):31. doi: 10.1186/1748-7188-8-31. Algorithms Mol Biol. 2013. PMID: 24350601 Free PMC article.
References
-
- Vassura M, Margara L, Di Lena P, Medri F, Fariselli P, et al. FT-COMAR: fault tolerant three-dimensional structure reconstruction from protein contact maps. Bioinformatics. 2008;24:1313–1315. - PubMed
-
- Vendruscolo M, Kussell E, Domany E. Recovery of protein structure from contact maps. Fold Des. 1997;2:295–306. - PubMed
-
- Mouradov D, Craven A, Forwood JK, Flanagan JU, Garcia-Castellanos R, et al. Modelling the structure of latexin-carboxypeptidase A complex based on chemical cross-linking and molecular docking. Protein Eng Des Sel. 2006;19:9–16. - PubMed
-
- Petrotchenko EV, Xiao K, Cable J, Chen Y, Dokholyan NV, et al. BiPS, a photo-cleavable, isotopically-coded, fluorescent crosslinker for structural proteomics. Mol Cell Proteomics 2008 - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
