

A proteomic view of an important human pathogen--towards the quantification of the entire Staphylococcus aureus proteome
- PMID: 19997597
- PMCID: PMC2781549
- DOI: 10.1371/journal.pone.0008176
A proteomic view of an important human pathogen--towards the quantification of the entire Staphylococcus aureus proteome
Abstract
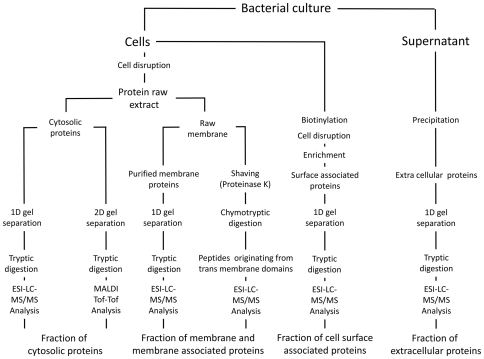
The genome sequence is the "blue-print of life," but proteomics provides the link to the actual physiology of living cells. Because of their low complexity bacteria are excellent model systems to identify the entire protein assembly of a living organism. Here we show that the majority of proteins expressed in growing and non-growing cells of the human pathogen Staphylococcus aureus can be identified and even quantified by a metabolic labeling proteomic approach. S. aureus has been selected as model for this proteomic study, because it poses a major risk to our health care system by combining high pathogenicity with an increasing frequency of multiple antibiotic resistance, thus requiring the development of new anti-staphylococcal therapy strategies. Since such strategies will likely have to target extracellular and surface-exposed virulence factors as well as staphylococcal survival and adaptation capabilities, we decided to combine four subproteomic fractions: cytosolic proteins, membrane-bound proteins, cell surface-associated and extracellular proteins, to comprehensively cover the entire proteome of S. aureus. This quantitative proteomics approach integrating data ranging from gene expression to subcellular localization in growing and non-growing cells is a proof of principle for whole-cell physiological proteomics that can now be extended to address physiological questions in infection-relevant settings. Importantly, with more than 1700 identified proteins (and 1450 quantified proteins) corresponding to a coverage of about three-quarters of the expressed proteins, our model study represents the most comprehensive quantification of a bacterial proteome reported to date. It thus paves the way towards a new level in understanding of cell physiology and pathophysiology of S. aureus and related pathogenic bacteria, opening new avenues for infection-related research on this crucial pathogen.
Conflict of interest statement
Figures







Similar articles
-
From the genome sequence via the proteome to cell physiology - Pathoproteomics and pathophysiology of Staphylococcus aureus.Int J Med Microbiol. 2018 Aug;308(6):545-557. doi: 10.1016/j.ijmm.2018.01.002. Epub 2018 Jan 5. Int J Med Microbiol. 2018. PMID: 29398252 Review.
-
Antibacterial mechanism of daptomycin antibiotic against Staphylococcus aureus based on a quantitative bacterial proteome analysis.J Proteomics. 2017 Jan 6;150:242-251. doi: 10.1016/j.jprot.2016.09.014. Epub 2016 Sep 29. J Proteomics. 2017. PMID: 27693894
-
Proteomics in studies of Staphylococcus aureus virulence.Acta Biochim Pol. 2015;62(3):367-81. doi: 10.18388/abp.2015_1083. Epub 2015 Aug 26. Acta Biochim Pol. 2015. PMID: 26307769 Review.
-
Proteomic approaches to study Staphylococcus aureus pathogenesis.J Proteomics. 2010 Feb 10;73(4):701-8. doi: 10.1016/j.jprot.2009.10.007. Epub 2009 Oct 29. J Proteomics. 2010. PMID: 19879388 Review.
-
A proteomic view of cell physiology and virulence of Staphylococcus aureus.Int J Med Microbiol. 2010 Feb;300(2-3):76-87. doi: 10.1016/j.ijmm.2009.10.006. Epub 2009 Dec 11. Int J Med Microbiol. 2010. PMID: 20005169 Review.
Cited by
-
Staphylococcus aureus sqr Encodes a Type II Sulfide:Quinone Oxidoreductase and Impacts Reactive Sulfur Speciation in Cells.Biochemistry. 2016 Nov 29;55(47):6524-6534. doi: 10.1021/acs.biochem.6b00714. Epub 2016 Nov 16. Biochemistry. 2016. PMID: 27806570 Free PMC article.
-
Proteomic profiling of the endogenous peptides of MRSA and MSSA.PeerJ. 2021 Nov 24;9:e12508. doi: 10.7717/peerj.12508. eCollection 2021. PeerJ. 2021. PMID: 34900427 Free PMC article.
-
The production of extracellular proteins is regulated by ribonuclease III via two different pathways in Staphylococcus aureus.PLoS One. 2011;6(5):e20554. doi: 10.1371/journal.pone.0020554. Epub 2011 May 31. PLoS One. 2011. PMID: 21655230 Free PMC article.
-
Activity control of the ClpC adaptor McsB in Bacillus subtilis.J Bacteriol. 2011 Aug;193(15):3887-93. doi: 10.1128/JB.00079-11. Epub 2011 May 27. J Bacteriol. 2011. PMID: 21622759 Free PMC article.
-
Omics Approaches for the Study of Adaptive Immunity to Staphylococcus aureus and the Selection of Vaccine Candidates.Proteomes. 2016 Mar 7;4(1):11. doi: 10.3390/proteomes4010011. Proteomes. 2016. PMID: 28248221 Free PMC article. Review.
References
-
- Boucher HW, Corey GR. Epidemiology of methicillin-resistant Staphylococcus aureus. Clin Infect Dis. 2008;46:344–9. - PubMed
-
- Wasinger VC, Cordwell SJ, Cerpa-Poljak A, Yan JX, Gooley AA, et al. Progress with gene-product mapping of the Mollicutes: Mycoplasma genitalium. Electrophoresis. 1995;7:1090–4. - PubMed
-
- Cravatt BF, Simon GM, Yates JR., 3rd The biological impact of mass-spectrometry-based proteomics. Nature. 2007;7172:991–1000. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
