

Whole-genome gene expression profiling of formalin-fixed, paraffin-embedded tissue samples
- PMID: 19997620
- PMCID: PMC2780295
- DOI: 10.1371/journal.pone.0008162
Whole-genome gene expression profiling of formalin-fixed, paraffin-embedded tissue samples
Abstract
Background: We have developed a gene expression assay (Whole-Genome DASL), capable of generating whole-genome gene expression profiles from degraded samples such as formalin-fixed, paraffin-embedded (FFPE) specimens.
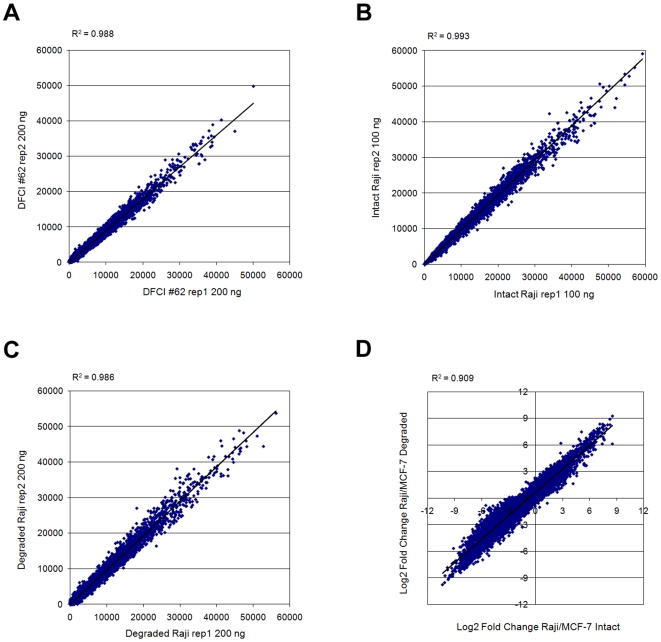
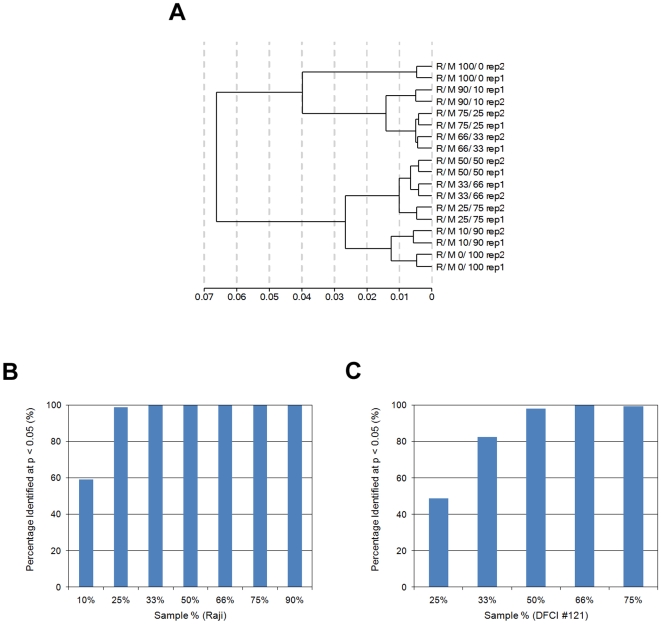
Methodology/principal findings: We demonstrated a similar level of sensitivity in gene detection between matched fresh-frozen (FF) and FFPE samples, with the number and overlap of probes detected in the FFPE samples being approximately 88% and 95% of that in the corresponding FF samples, respectively; 74% of the differentially expressed probes overlapped between the FF and FFPE pairs. The WG-DASL assay is also able to detect 1.3-1.5 and 1.5-2 -fold changes in intact and FFPE samples, respectively. The dynamic range for the assay is approximately 3 logs. Comparing the WG-DASL assay with an in vitro transcription-based labeling method yielded fold-change correlations of R(2) approximately 0.83, while fold-change comparisons with quantitative RT-PCR assays yielded R(2) approximately 0.86 and R(2) approximately 0.55 for intact and FFPE samples, respectively. Additionally, the WG-DASL assay yielded high self-correlations (R(2)>0.98) with low intact RNA inputs ranging from 1 ng to 100 ng; reproducible expression profiles were also obtained with 250 pg total RNA (R(2) approximately 0.92), with approximately 71% of the probes detected in 100 ng total RNA also detected at the 250 pg level. When FFPE samples were assayed, 1 ng total RNA yielded self-correlations of R(2) approximately 0.80, while still maintaining a correlation of R(2) approximately 0.75 with standard FFPE inputs (200 ng).
Conclusions/significance: Taken together, these results show that WG-DASL assay provides a reliable platform for genome-wide expression profiling in archived materials. It also possesses utility within clinical settings where only limited quantities of samples may be available (e.g. microdissected material) or when minimally invasive procedures are performed (e.g. biopsied specimens).
Conflict of interest statement
Figures



Similar articles
-
Analyses and interpretation of whole-genome gene expression from formalin-fixed paraffin-embedded tissue: an illustration with breast cancer tissues.BMC Genomics. 2010 Nov 8;11:622. doi: 10.1186/1471-2164-11-622. BMC Genomics. 2010. PMID: 21059268 Free PMC article.
-
Gene expression profiles from formalin fixed paraffin embedded breast cancer tissue are largely comparable to fresh frozen matched tissue.PLoS One. 2011 Feb 11;6(2):e17163. doi: 10.1371/journal.pone.0017163. PLoS One. 2011. PMID: 21347257 Free PMC article.
-
Reliable gene expression profiling of formalin-fixed paraffin-embedded breast cancer tissue (FFPE) using cDNA-mediated annealing, extension, selection, and ligation whole-genome (DASL WG) assay.BMC Med Genomics. 2016 Aug 20;9(1):54. doi: 10.1186/s12920-016-0215-4. BMC Med Genomics. 2016. PMID: 27542606 Free PMC article.
-
Accuracy of Molecular Data Generated with FFPE Biospecimens: Lessons from the Literature.Cancer Res. 2015 Apr 15;75(8):1541-7. doi: 10.1158/0008-5472.CAN-14-2378. Epub 2015 Apr 2. Cancer Res. 2015. PMID: 25836717 Free PMC article. Review.
-
Systematic review and feasibility study on pre-analytical factors and genomic analyses on archival formalin-fixed paraffin-embedded breast cancer tissue.Sci Rep. 2024 Aug 6;14(1):18275. doi: 10.1038/s41598-024-69285-8. Sci Rep. 2024. PMID: 39107471 Free PMC article.
Cited by
-
An Integrated Transcriptomic Approach to Identify Molecular Markers of Calcineurin Inhibitor Nephrotoxicity in Pediatric Kidney Transplant Recipients.Int J Mol Sci. 2021 May 21;22(11):5414. doi: 10.3390/ijms22115414. Int J Mol Sci. 2021. PMID: 34063776 Free PMC article.
-
A Comparison of Fresh Frozen vs. Formalin-Fixed, Paraffin-Embedded Specimens of Canine Mammary Tumors via Branched-DNA Assay.Int J Mol Sci. 2016 May 13;17(5):724. doi: 10.3390/ijms17050724. Int J Mol Sci. 2016. PMID: 27187374 Free PMC article.
-
A microRNA activity map of human mesenchymal tumors: connections to oncogenic pathways; an integrative transcriptomic study.BMC Genomics. 2012 Jul 23;13:332. doi: 10.1186/1471-2164-13-332. BMC Genomics. 2012. PMID: 22823907 Free PMC article.
-
Potential prognostic biomarkers for bone metastasis from hepatocellular carcinoma.Oncologist. 2011;16(7):1028-39. doi: 10.1634/theoncologist.2010-0358. Epub 2011 Jun 10. Oncologist. 2011. PMID: 21665914 Free PMC article.
-
Coextraction and PCR Based Analysis of Nucleic Acids From Formalin-Fixed Paraffin-Embedded Specimens.J Clin Lab Anal. 2015 Nov;29(6):485-92. doi: 10.1002/jcla.21798. Epub 2014 Oct 2. J Clin Lab Anal. 2015. PMID: 25277467 Free PMC article.
References
-
- Bouchie A. Coming soon: a global grid for cancer research. Nat Biotechnol. 2004;22:1071–1073. - PubMed
-
- Ramaswamy S. Translating cancer genomics into clinical oncology. N Engl J Med. 2004;350:1814–1816. - PubMed
-
- Farragher SM, Tanney A, Kennedy RD, Paul Harkin D. RNA expression analysis from formalin fixed paraffin embedded tissues. Histochem Cell Biol. 2008;130:435–445. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
