

De novo structure generation using chemical shifts for proteins with high-sequence identity but different folds
- PMID: 19998407
- PMCID: PMC2865713
- DOI: 10.1002/pro.303
De novo structure generation using chemical shifts for proteins with high-sequence identity but different folds
Abstract
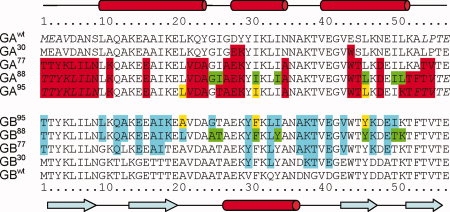
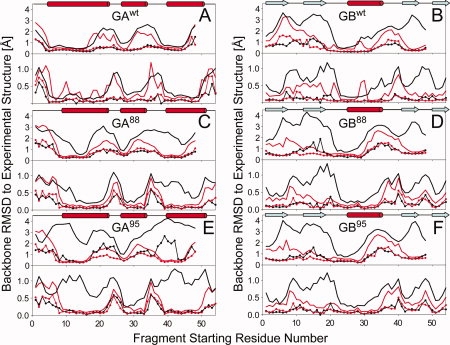
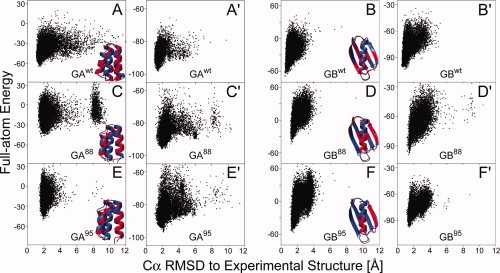
Proteins with high-sequence identity but very different folds present a special challenge to sequence-based protein structure prediction methods. In particular, a 56-residue three-helical bundle protein (GA(95)) and an alpha/beta-fold protein (GB(95)), which share 95% sequence identity, were targets in the CASP-8 structure prediction contest. With only 12 out of 300 submitted server-CASP8 models for GA(95) exhibiting the correct fold, this protein proved particularly challenging despite its small size. Here, we demonstrate that the information contained in NMR chemical shifts can readily be exploited by the CS-Rosetta structure prediction program and yields adequate convergence, even when input chemical shifts are limited to just amide (1)H(N) and (15)N or (1)H(N) and (1)H(alpha) values.
Figures
References
-
- Burley SK. An overview of structural genomics. Nat Struct Biol. 2000;7:932–934. - PubMed
-
- Marti-Renom MA, Stuart AC, Fiser A, Sanchez R, Melo F, Sali A. Comparative protein structure modeling of genes and genomes. Annu Rev Biophys Biomol Struct. 2000;29:291–325. - PubMed
-
- Domingues FS, Lackner P, Andreeva A, Sippl MJ. Structure-based evaluation of sequence comparison and fold recognition alignment accuracy. J Mol Biol. 2000;297:1003–1013. - PubMed
-
- Baker D, Sali A. Protein structure prediction and structural genomics. Science. 2001;294:93–96. - PubMed
-
- Das R, Baker D. Macromolecular modeling with Rosetta. Annu Rev Biochem. 2008;77:363–382. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
