

Potential therapeutic benefits of strategies directed to mitochondria
- PMID: 20001744
- PMCID: PMC2936955
- DOI: 10.1089/ars.2009.2788
Potential therapeutic benefits of strategies directed to mitochondria
Abstract
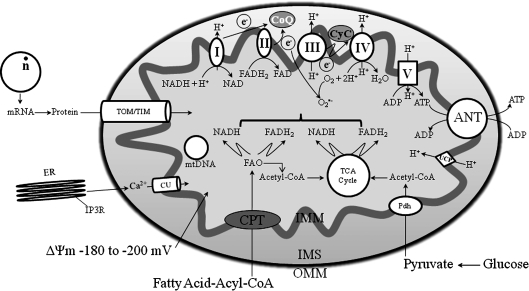
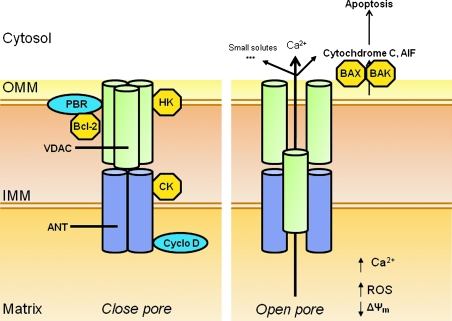
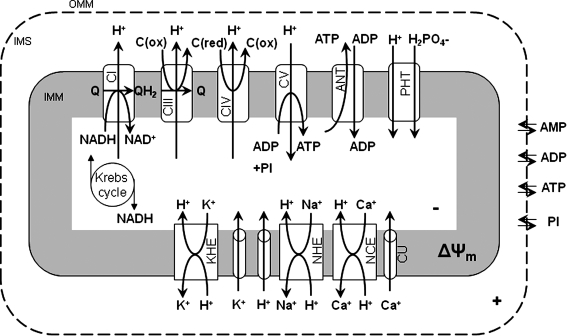
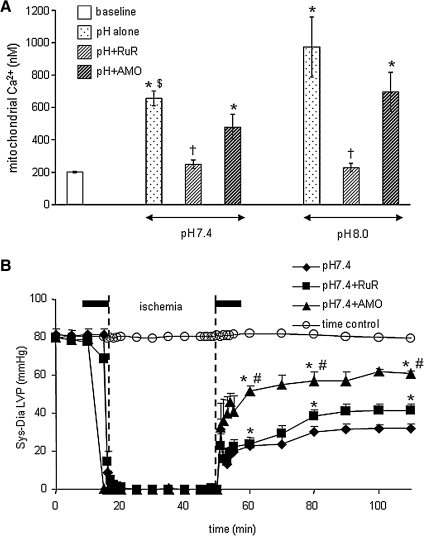
The mitochondrion is the most important organelle in determining continued cell survival and cell death. Mitochondrial dysfunction leads to many human maladies, including cardiovascular diseases, neurodegenerative disease, and cancer. These mitochondria-related pathologies range from early infancy to senescence. The central premise of this review is that if mitochondrial abnormalities contribute to the pathological state, alleviating the mitochondrial dysfunction would contribute to attenuating the severity or progression of the disease. Therefore, this review will examine the role of mitochondria in the etiology and progression of several diseases and explore potential therapeutic benefits of targeting mitochondria in mitigating the disease processes. Indeed, recent advances in mitochondrial biology have led to selective targeting of drugs designed to modulate and manipulate mitochondrial function and genomics for therapeutic benefit. These approaches to treat mitochondrial dysfunction rationally could lead to selective protection of cells in different tissues and various disease states. However, most of these approaches are in their infancy.
Figures
Similar articles
-
Mitochondrial approaches to protect against cardiac ischemia and reperfusion injury.Front Physiol. 2011 Apr 12;2:13. doi: 10.3389/fphys.2011.00013. eCollection 2011. Front Physiol. 2011. PMID: 21559063 Free PMC article.
-
[Mitochondria in cell life, death and disease].Postepy Biochem. 2008;54(2):129-41. Postepy Biochem. 2008. PMID: 18807924 Review. Polish.
-
Melatonin-mitochondria interplay in health and disease.Curr Top Med Chem. 2011;11(2):221-40. doi: 10.2174/156802611794863517. Curr Top Med Chem. 2011. PMID: 21244359 Review.
-
Mitochondria-directed therapeutics.Antioxid Redox Signal. 2008 Mar;10(3):575-8. doi: 10.1089/ars.2007.1929. Antioxid Redox Signal. 2008. PMID: 18092938
-
Mitochondria: a hub of redox activities and cellular distress control.Mol Cell Biochem. 2007 Nov;305(1-2):235-53. doi: 10.1007/s11010-007-9520-8. Epub 2007 Jun 12. Mol Cell Biochem. 2007. PMID: 17562131 Review.
Cited by
-
Impaired Myocardial Mitochondrial Function in an Experimental Model of Anaphylactic Shock.Biology (Basel). 2022 May 10;11(5):730. doi: 10.3390/biology11050730. Biology (Basel). 2022. PMID: 35625458 Free PMC article.
-
Monitoring mitochondrial electron fluxes using NAD(P)H-flavoprotein fluorometry reveals complex action of isoflurane on cardiomyocytes.Biochim Biophys Acta. 2010 Oct;1797(10):1749-58. doi: 10.1016/j.bbabio.2010.07.009. Epub 2010 Jul 17. Biochim Biophys Acta. 2010. PMID: 20646994 Free PMC article.
-
Hypothermia Prevents Cardiac Dysfunction during Acute Ischemia Reperfusion by Maintaining Mitochondrial Bioenergetics and by Promoting Hexokinase II Binding to Mitochondria.Oxid Med Cell Longev. 2022 Jul 13;2022:4476448. doi: 10.1155/2022/4476448. eCollection 2022. Oxid Med Cell Longev. 2022. PMID: 35873800 Free PMC article.
-
Alpha-linolenic acid stabilizes HIF-1 α and downregulates FASN to promote mitochondrial apoptosis for mammary gland chemoprevention.Oncotarget. 2017 Jul 25;8(41):70049-70071. doi: 10.18632/oncotarget.19551. eCollection 2017 Sep 19. Oncotarget. 2017. PMID: 29050261 Free PMC article.
-
Tyrosine nitration of voltage-dependent anion channels in cardiac ischemia-reperfusion: reduction by peroxynitrite scavenging.Biochim Biophys Acta. 2012 Nov;1817(11):2049-59. doi: 10.1016/j.bbabio.2012.06.004. Epub 2012 Jun 15. Biochim Biophys Acta. 2012. PMID: 22709907 Free PMC article.
References
-
- Abete P. Testa G. Ferrara N. De Santis D. Capaccio P. Viati L. Calabrese C. Cacciatore F. Longobardi G. Condorelli M. Napoli C. Rengo F. Cardioprotective effect of ischemic preconditioning is preserved in food-restricted senescent rats. Am J Physiol Heart Circ Physiol. 2002;282:H1978–H1987. - PubMed
-
- Abu–Hamad S. Zaid H. Israelson A. Nahon E. Shoshan–Barmatz V. Hexokinase-I protection against apoptotic cell death is mediated via interaction with the voltage-dependent anion channel-1: Mapping the site of binding. J Biol Chem. 2008;283:13482–13490. - PubMed
-
- Adams JM. Cory S. The Bcl-2 protein family: Arbiters of cell survival. Science. 1998;281:1322–1326. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
