

Post-transcriptional regulation of MEK-1 by polyamines through the RNA-binding protein HuR modulating intestinal epithelial apoptosis
- PMID: 20001965
- PMCID: PMC3021782
- DOI: 10.1042/BJ20091459
Post-transcriptional regulation of MEK-1 by polyamines through the RNA-binding protein HuR modulating intestinal epithelial apoptosis
Abstract
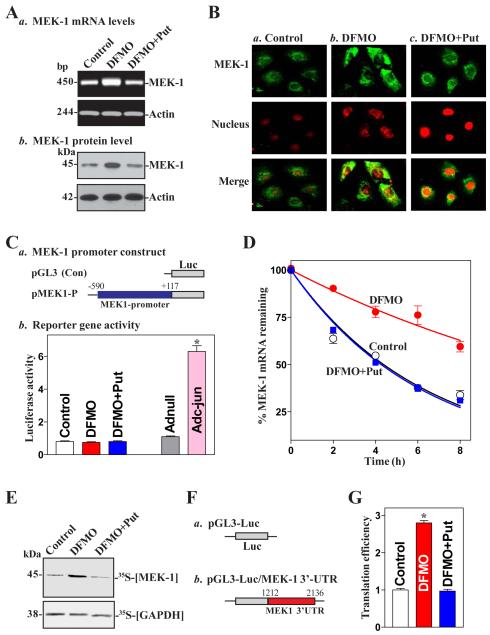
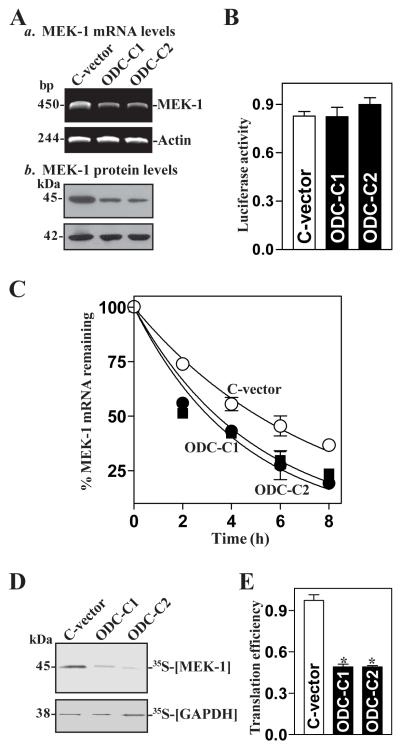
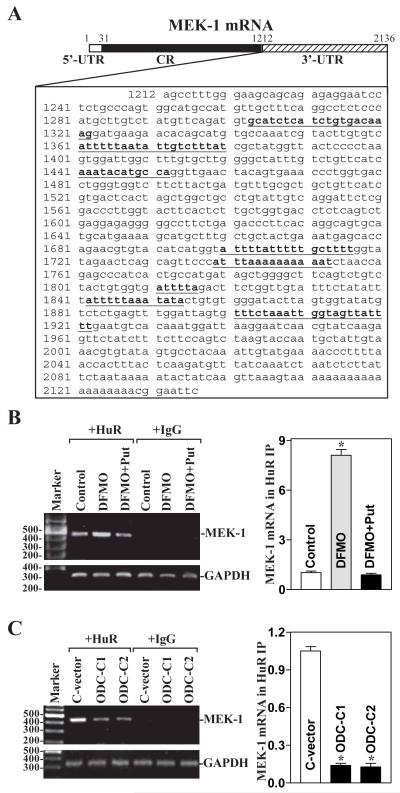
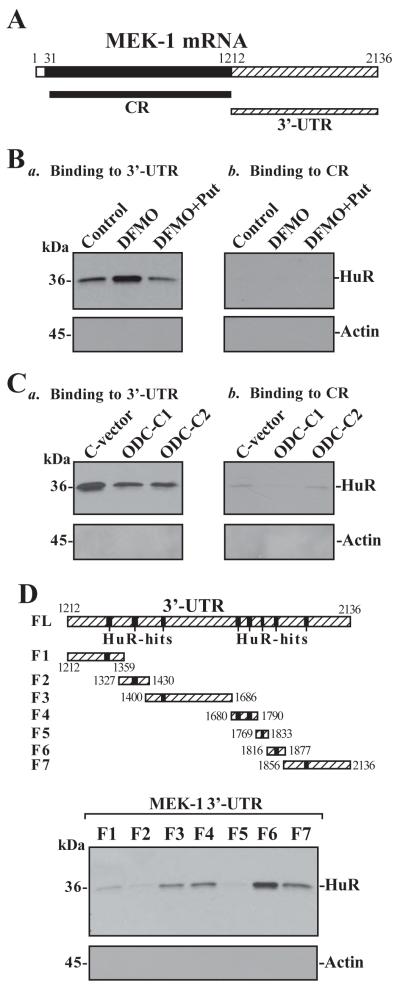
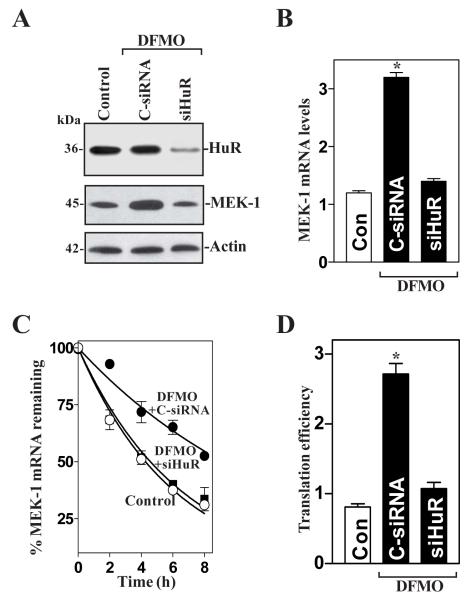
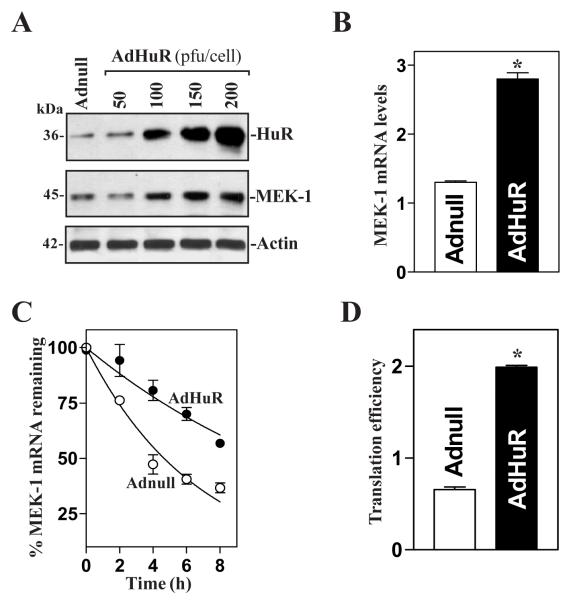
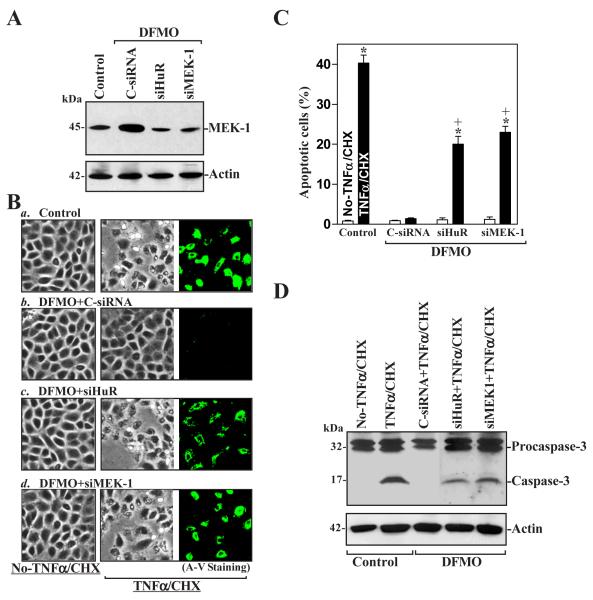
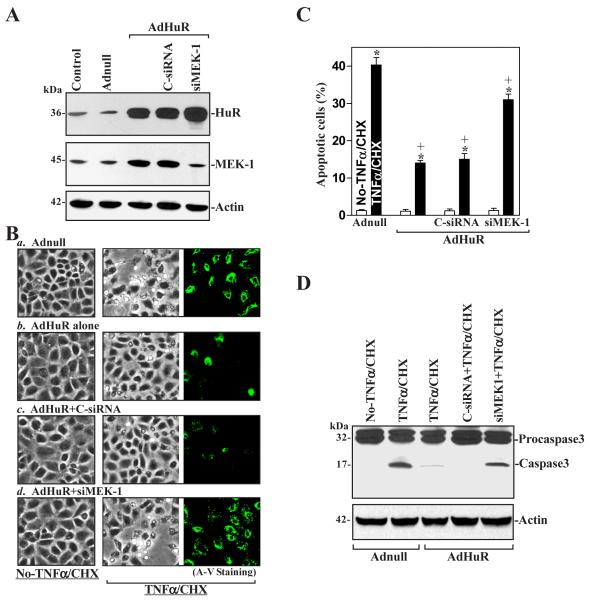
MEK-1 [MAPK (mitogen-activated protein kinase) kinase-1] is an important signal transducing enzyme that is implicated in many aspects of cellular functions. In the present paper, we report that cellular polyamines regulate MEK-1 expression at the post-transcriptional level through the RNA-binding protein HuR (Hu-antigen R) in IECs (intestinal epithelial cells). Decreasing the levels of cellular polyamines by inhibiting ODC (ornithine decarboxylase) stabilized MEK-1 mRNA and promoted its translation through enhancement of the interaction between HuR and the 3'-untranslated region of MEK-1 mRNA, whereas increasing polyamine levels by ectopic ODC overexpression destabilized the MEK-1 transcript and repressed its translation by reducing the abundance of HuR-MEK-1 mRNA complex; neither intervention changed MEK-1 gene transcription via its promoter. HuR silencing rendered the MEK-1 mRNA unstable and inhibited its translation, thus preventing increases in MEK-1 mRNA and protein in polyamine-deficient cells. Conversely, HuR overexpression increased MEK-1 mRNA stability and promoted its translation. Inhibition of MEK-1 expression by MEK-1 silencing or HuR silencing prevented the increased resistance of polyamine-deficient cells to apoptosis. Moreover, HuR overexpression did not protect against apoptosis if MEK-1 expression was silenced. These results indicate that polyamines destabilize the MEK-1 mRNA and repress its translation by inhibiting the association between HuR and the MEK-1 transcript. Our findings indicate that MEK-1 is a key effector of the HuR-elicited anti-apoptotic programme in IECs.
Figures
Similar articles
-
Polyamines regulate the stability of JunD mRNA by modulating the competitive binding of its 3' untranslated region to HuR and AUF1.Mol Cell Biol. 2010 Nov;30(21):5021-32. doi: 10.1128/MCB.00807-10. Epub 2010 Aug 30. Mol Cell Biol. 2010. PMID: 20805360 Free PMC article.
-
Polyamines regulate the stability of activating transcription factor-2 mRNA through RNA-binding protein HuR in intestinal epithelial cells.Mol Biol Cell. 2007 Nov;18(11):4579-90. doi: 10.1091/mbc.e07-07-0675. Epub 2007 Sep 5. Mol Biol Cell. 2007. PMID: 17804813 Free PMC article.
-
Stabilization of XIAP mRNA through the RNA binding protein HuR regulated by cellular polyamines.Nucleic Acids Res. 2009 Dec;37(22):7623-37. doi: 10.1093/nar/gkp755. Nucleic Acids Res. 2009. PMID: 19825980 Free PMC article.
-
HuR and mRNA stability.Cell Mol Life Sci. 2001 Feb;58(2):266-77. doi: 10.1007/PL00000854. Cell Mol Life Sci. 2001. PMID: 11289308 Free PMC article. Review.
-
HuR function in disease.Front Biosci (Landmark Ed). 2012 Jan 1;17(1):189-205. doi: 10.2741/3921. Front Biosci (Landmark Ed). 2012. PMID: 22201738 Free PMC article. Review.
Cited by
-
RNA-binding protein HuR promotes growth of small intestinal mucosa by activating the Wnt signaling pathway.Mol Biol Cell. 2014 Nov 1;25(21):3308-18. doi: 10.1091/mbc.E14-03-0853. Epub 2014 Aug 27. Mol Biol Cell. 2014. PMID: 25165135 Free PMC article.
-
Sm-site containing mRNAs can accept Sm-rings and are downregulated in Spinal Muscular Atrophy.Nucleic Acids Res. 2025 Aug 11;53(15):gkaf794. doi: 10.1093/nar/gkaf794. Nucleic Acids Res. 2025. PMID: 40823813 Free PMC article.
-
Polyamines regulate the stability of JunD mRNA by modulating the competitive binding of its 3' untranslated region to HuR and AUF1.Mol Cell Biol. 2010 Nov;30(21):5021-32. doi: 10.1128/MCB.00807-10. Epub 2010 Aug 30. Mol Cell Biol. 2010. PMID: 20805360 Free PMC article.
-
Inhibition of Smurf2 translation by miR-322/503 modulates TGF-β/Smad2 signaling and intestinal epithelial homeostasis.Mol Biol Cell. 2014 Apr;25(8):1234-43. doi: 10.1091/mbc.E13-09-0560. Epub 2014 Feb 19. Mol Biol Cell. 2014. PMID: 24554769 Free PMC article.
-
miR-195 competes with HuR to modulate stim1 mRNA stability and regulate cell migration.Nucleic Acids Res. 2013 Sep;41(16):7905-19. doi: 10.1093/nar/gkt565. Epub 2013 Jun 26. Nucleic Acids Res. 2013. PMID: 23804758 Free PMC article.
References
-
- Radtke F, Clevers H. Self-renewal and cancer of the gut: two sides of a coin. Science. 2005;307:1904–1909. - PubMed
-
- Wang JY. Polyamines and mRNA stability in regulation of intestinal mucosal growth. Amino. Acids. 2005;33:241–252. - PubMed
-
- Orphanides G, Reinberg D. A unified theory of gene expression. Cell. 2002;108:439–451. - PubMed
-
- Li L, Liu L, Rao JN, Esmaili A, Strauch ED, Bass BL, Wang JY. JunD stabilization results in inhibition of normal intestinal epithelial cell growth through p21 after polyamine depletion. Gastroenterology. 2002;123:764–779. - PubMed
-
- Li L, Rao JN, Guo X, Liu L, Santora R, Bass BL, Wang JY. Polyamine depletion stabilizes p53 resulting in inhibition of normal intestinal epithelial cell proliferation. Am. J. Physiol. Cell. Physiol. 2001;281:C941–C953. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous