

Crude subcellular fractionation of cultured mammalian cell lines
- PMID: 20003239
- PMCID: PMC2802353
- DOI: 10.1186/1756-0500-2-243
Crude subcellular fractionation of cultured mammalian cell lines
Abstract
Background: The expression and study of recombinant proteins in mammalian culture systems can be complicated during the cell lysis procedure by contaminating proteins from cellular compartments distinct from those within which the protein of interest resides and also by solubility issues that may arise from the use of a single lysis buffer. Partial subcellular fractionation using buffers of increasing stringency, rather than whole cell lysis is one way in which to avoid or reduce this contamination and ensure complete recovery of the target protein. Currently published protocols involve time consuming centrifugation steps which may require expensive equipment and commercially available kits can be prohibitively expensive when handling large or multiple samples.
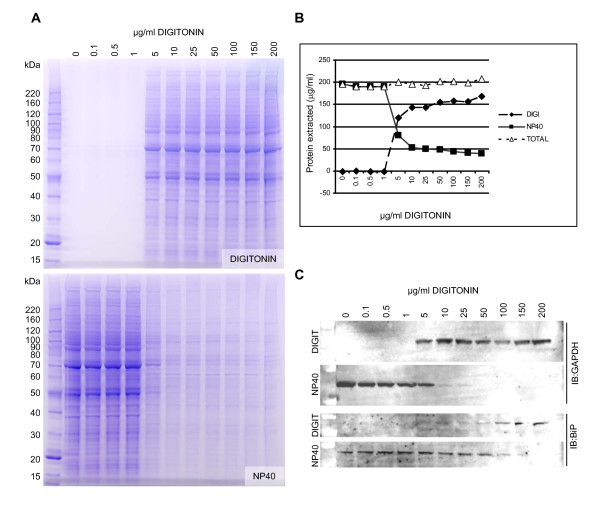
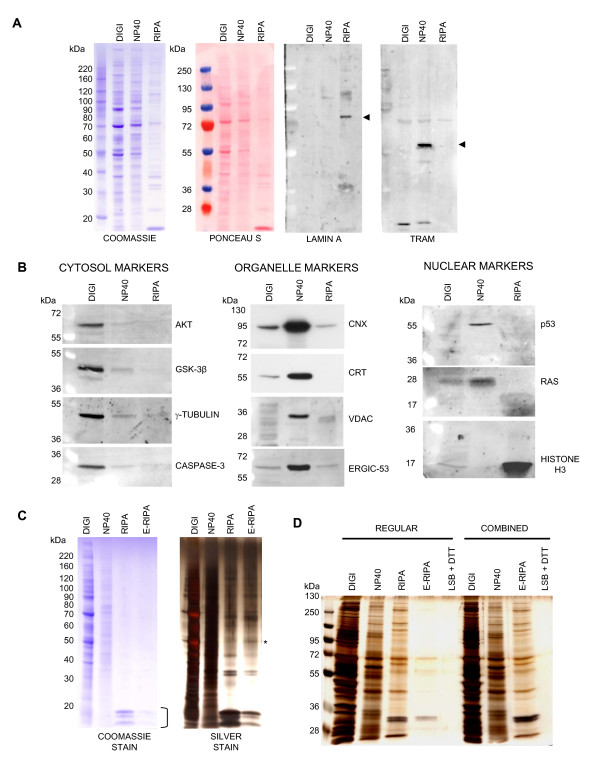
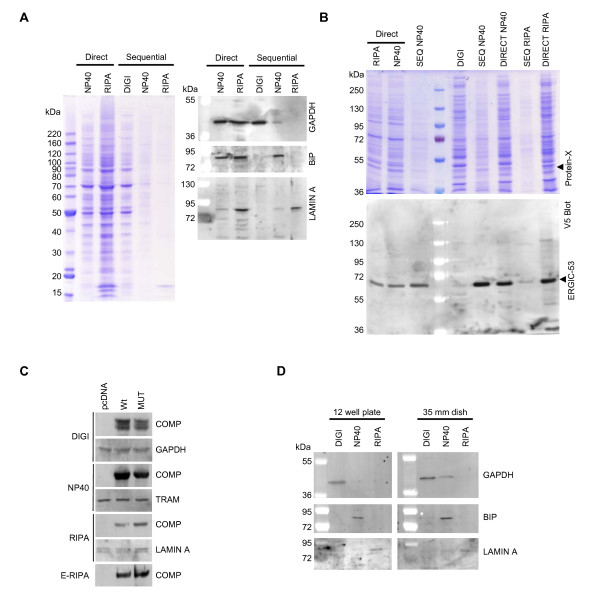
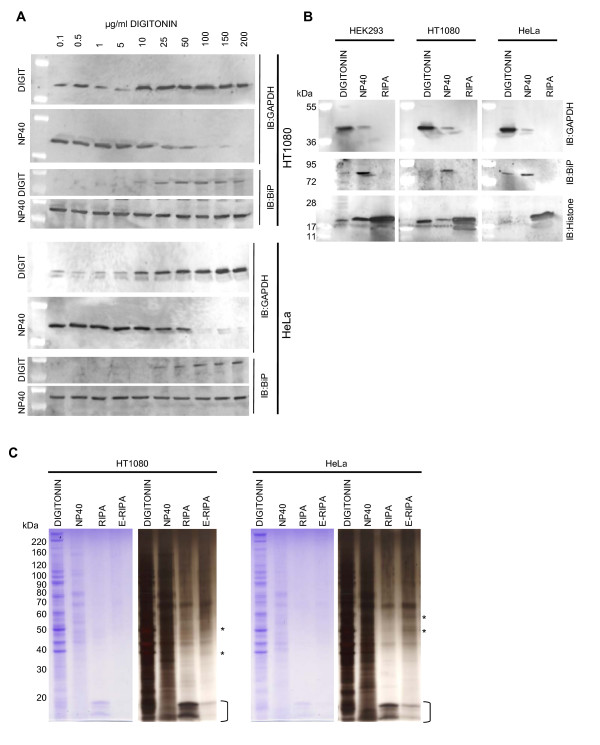
Findings: We have established a protocol to sequentially extract proteins from cultured mammalian cells in fractions enriched for cytosolic, membrane bound organellar, nuclear and insoluble proteins. All of the buffers used can be made inexpensively and easily and the protocol requires no costly equipment. While the method was optimized for a specific cell type, we demonstrate that the protocol can be applied to a variety of commonly used cell lines and anticipate that it can be applied to any cell line via simple optimization of the primary extraction step.
Conclusion: We describe a protocol for the crude subcellular fractionation of cultured mammalian cells that is both straightforward and cost effective and may facilitate the more accurate study of recombinant proteins and the generation of purer preparations of said proteins from cell extracts.
Figures

References
LinkOut - more resources
Full Text Sources
Other Literature Sources
