

An efficient algorithm for the stochastic simulation of the hybridization of DNA to microarrays
- PMID: 20003312
- PMCID: PMC2805644
- DOI: 10.1186/1471-2105-10-411
An efficient algorithm for the stochastic simulation of the hybridization of DNA to microarrays
Abstract
Background: Although oligonucleotide microarray technology is ubiquitous in genomic research, reproducibility and standardization of expression measurements still concern many researchers. Cross-hybridization between microarray probes and non-target ssDNA has been implicated as a primary factor in sensitivity and selectivity loss. Since hybridization is a chemical process, it may be modeled at a population-level using a combination of material balance equations and thermodynamics. However, the hybridization reaction network may be exceptionally large for commercial arrays, which often possess at least one reporter per transcript. Quantification of the kinetics and equilibrium of exceptionally large chemical systems of this type is numerically infeasible with customary approaches.
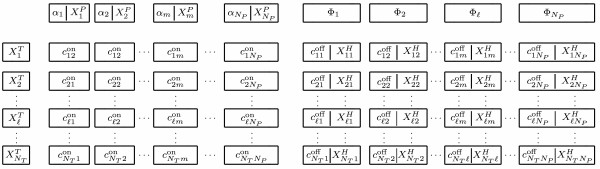
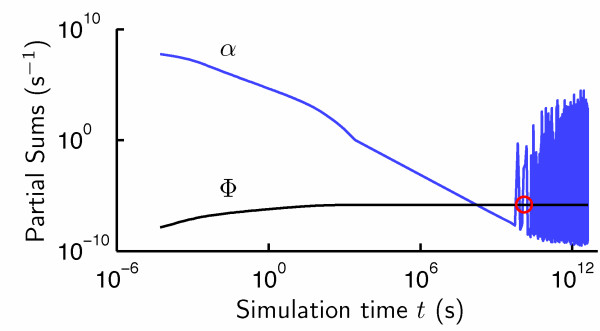
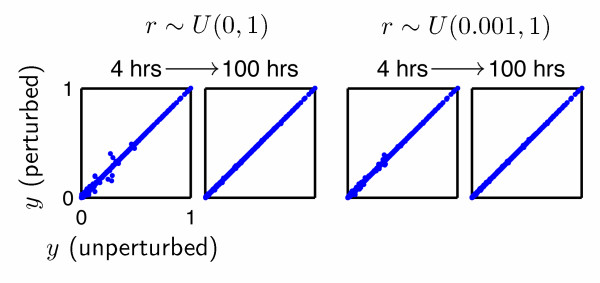
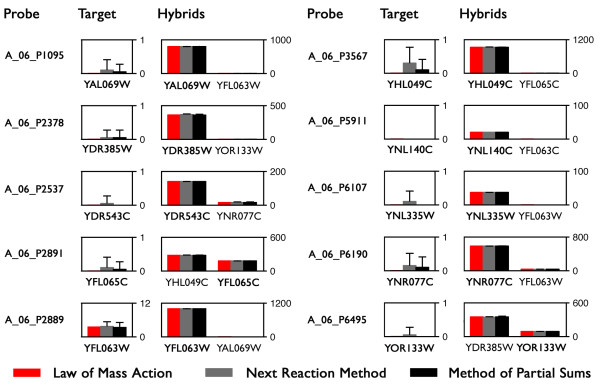
Results: In this paper, we present a robust and computationally efficient algorithm for the simulation of hybridization processes underlying microarray assays. Our method may be utilized to identify the extent to which nucleic acid targets (e.g. cDNA) will cross-hybridize with probes, and by extension, characterize probe robustnessusing the information specified by MAGE-TAB. Using this algorithm, we characterize cross-hybridization in a modified commercial microarray assay.
Conclusions: By integrating stochastic simulation with thermodynamic prediction tools for DNA hybridization, one may robustly and rapidly characterize of the selectivity of a proposed microarray design at the probe and "system" levels. Our code is available at http://www.laurenzi.net.
Figures
Similar articles
-
UPS 2.0: unique probe selector for probe design and oligonucleotide microarrays at the pangenomic/genomic level.BMC Genomics. 2010 Dec 2;11 Suppl 4(Suppl 4):S6. doi: 10.1186/1471-2164-11-S4-S6. BMC Genomics. 2010. PMID: 21143815 Free PMC article.
-
Transcript mapping with high-density oligonucleotide tiling arrays.Bioinformatics. 2006 Aug 15;22(16):1963-70. doi: 10.1093/bioinformatics/btl289. Epub 2006 Jun 20. Bioinformatics. 2006. PMID: 16787969
-
A generic approach for the design of whole-genome oligoarrays, validated for genomotyping, deletion mapping and gene expression analysis on Staphylococcus aureus.BMC Genomics. 2005 Jun 17;6:95. doi: 10.1186/1471-2164-6-95. BMC Genomics. 2005. PMID: 15963225 Free PMC article.
-
Genomic profiling: cDNA arrays and oligoarrays.Methods Mol Biol. 2012;823:89-105. doi: 10.1007/978-1-60327-216-2_7. Methods Mol Biol. 2012. PMID: 22081341 Review.
-
DNA hybridization arrays for gene expression analysis of human oral cancer.J Dent Res. 2002 Feb;81(2):89-97. J Dent Res. 2002. PMID: 11829015 Review.
Cited by
-
Development and evaluation of a new detection tool-visual DNA microarray for simultaneous and specific detection of human immunodeficiency virus type-1 and hepatitis C virus.Mol Biol Rep. 2011 Nov;38(8):5341-8. doi: 10.1007/s11033-011-0685-6. Epub 2011 Feb 26. Mol Biol Rep. 2011. PMID: 21359643
-
Nucleic acid functionalized fiber optic probes for sensing in evanescent wave: optimization and application.RSC Adv. 2019 Jan 18;9(4):2316-2324. doi: 10.1039/c8ra10125f. eCollection 2019 Jan 14. RSC Adv. 2019. PMID: 35516110 Free PMC article.
References
-
- Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, Berka J, Braverman MS, Chen YJ, Chen Z, Dewell SB, Du L, Fierro JM, Gomes XV, Godwin BC, He W, Helgesen S, Ho CH, Irzyk GP, Jando SC, Alenquer MLI, Jarvie TP, Jirage KB, Kim JB, Knight JR, Lanza JR, Leamon JH, Lefkowitz SM, Lei M, Li J, Lohman KL, Lu H, Makhijani VB, McDade KE, McKenna MP, Myers EW, Nickerson E, Nobile JR, Plant R, Puc BP, Ronan MT, Roth GT, Sarkis GJ, Simons JF, Simpson JW, Srinivasan M, Tartaro KR, Tomasz A, Vogt KA, Volkmer GA, Wang SH, Wang Y, Weiner MP, Yu P, Begley RF, Rothberg JM. Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005;437:376–380. - PMC - PubMed
-
- Cloonan N, Forrest ARR, Kolle G, Gardiner BBA, Faulkner GJ, Brown MK, Taylor DF, Steptoe AL, Wani S, Bethel G, Robertson AJ, Perkins AC, Bruce SJ, Lee CC, Ranade SS, Peckham HE, Manning JM, McKernan KJ, Grimmond SM. Stem cell transcriptome profiling via massive-scale mRNA sequencing. Nature Methods. 2008;5:613–619. doi: 10.1038/nmeth.1223. - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
