

Modular prediction of protein structural classes from sequences of twilight-zone identity with predicting sequences
- PMID: 20003388
- PMCID: PMC2805645
- DOI: 10.1186/1471-2105-10-414
Modular prediction of protein structural classes from sequences of twilight-zone identity with predicting sequences
Abstract
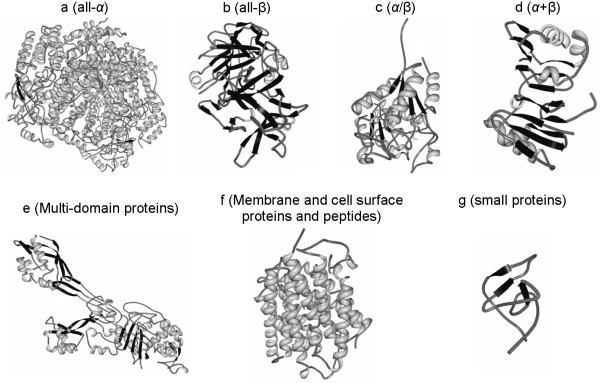
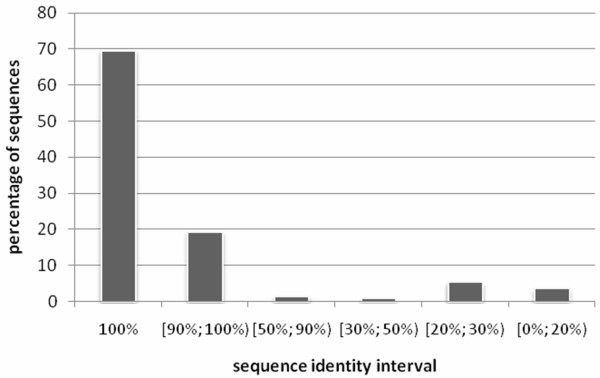
Background: Knowledge of structural class is used by numerous methods for identification of structural/functional characteristics of proteins and could be used for the detection of remote homologues, particularly for chains that share twilight-zone similarity. In contrast to existing sequence-based structural class predictors, which target four major classes and which are designed for high identity sequences, we predict seven classes from sequences that share twilight-zone identity with the training sequences.
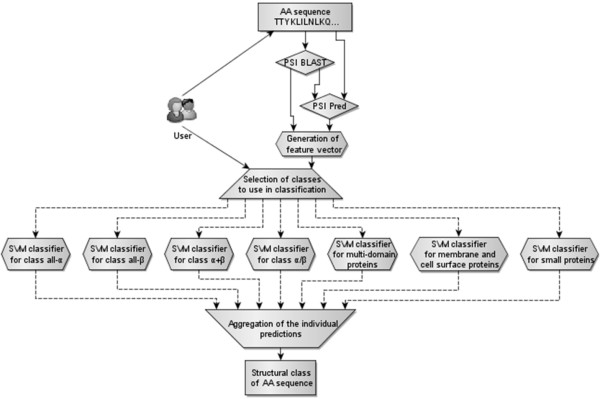
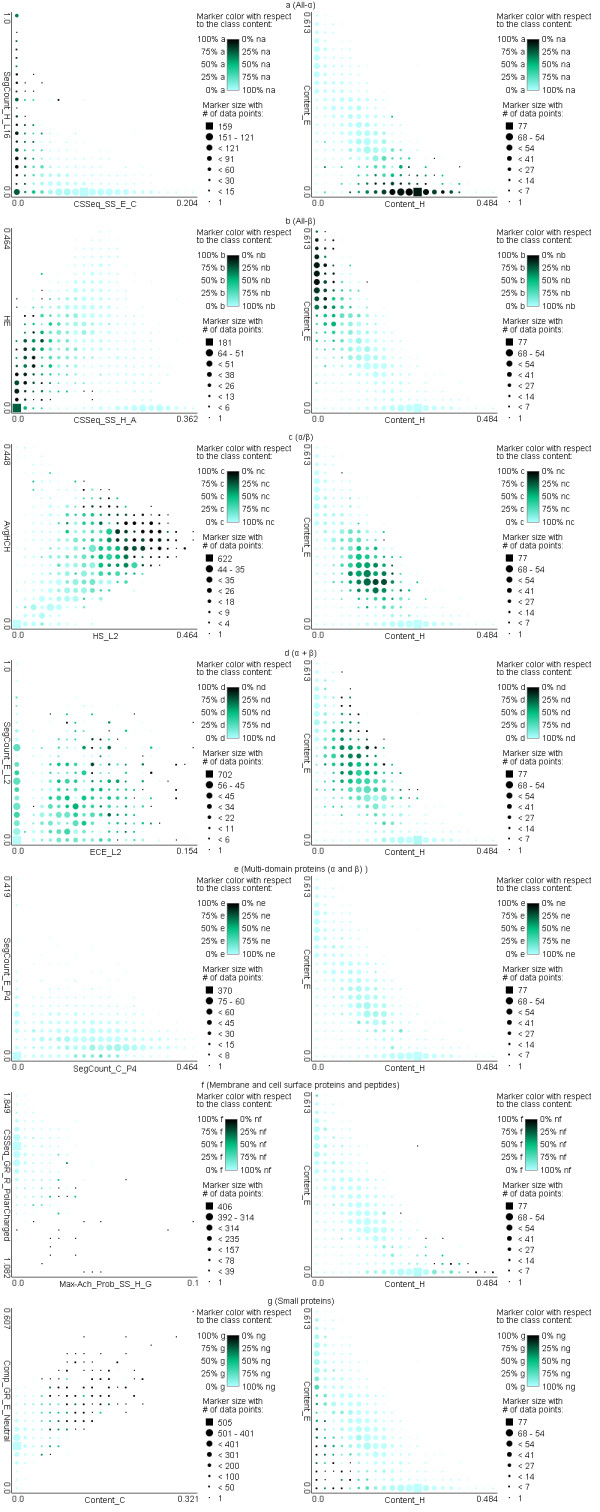
Results: The proposed MODular Approach to Structural class prediction (MODAS) method is unique as it allows for selection of any subset of the classes. MODAS is also the first to utilize a novel, custom-built feature-based sequence representation that combines evolutionary profiles and predicted secondary structure. The features quantify information relevant to the definition of the classes including conservation of residues and arrangement and number of helix/strand segments. Our comprehensive design considers 8 feature selection methods and 4 classifiers to develop Support Vector Machine-based classifiers that are tailored for each of the seven classes. Tests on 5 twilight-zone and 1 high-similarity benchmark datasets and comparison with over two dozens of modern competing predictors show that MODAS provides the best overall accuracy that ranges between 80% and 96.7% (83.5% for the twilight-zone datasets), depending on the dataset. This translates into 19% and 8% error rate reduction when compared against the best performing competing method on two largest datasets. The proposed predictor provides accurate predictions at 58% accuracy for membrane proteins class, which is not considered by majority of existing methods, in spite that this class accounts for only 2% of the data. Our predictive model is analyzed to demonstrate how and why the input features are associated with the corresponding classes.
Conclusions: The improved predictions stem from the novel features that express collocation of the secondary structure segments in the protein sequence and that combine evolutionary and secondary structure information. Our work demonstrates that conservation and arrangement of the secondary structure segments predicted along the protein chain can successfully predict structural classes which are defined based on the spatial arrangement of the secondary structures. A web server is available at http://biomine.ece.ualberta.ca/MODAS/.
Figures
Similar articles
-
SCPRED: accurate prediction of protein structural class for sequences of twilight-zone similarity with predicting sequences.BMC Bioinformatics. 2008 May 1;9:226. doi: 10.1186/1471-2105-9-226. BMC Bioinformatics. 2008. PMID: 18452616 Free PMC article.
-
Prediction of protein structural class using novel evolutionary collocation-based sequence representation.J Comput Chem. 2008 Jul 30;29(10):1596-604. doi: 10.1002/jcc.20918. J Comput Chem. 2008. PMID: 18293306
-
Prediction of beta-turns at over 80% accuracy based on an ensemble of predicted secondary structures and multiple alignments.BMC Bioinformatics. 2008 Oct 10;9:430. doi: 10.1186/1471-2105-9-430. BMC Bioinformatics. 2008. PMID: 18847492 Free PMC article.
-
General overview on structure prediction of twilight-zone proteins.Theor Biol Med Model. 2015 Sep 4;12:15. doi: 10.1186/s12976-015-0014-1. Theor Biol Med Model. 2015. PMID: 26338054 Free PMC article. Review.
-
Prediction of protein structural class based on symmetrical recurrence quantification analysis.Comput Biol Chem. 2021 Jun;92:107450. doi: 10.1016/j.compbiolchem.2021.107450. Epub 2021 Feb 8. Comput Biol Chem. 2021. PMID: 33631460 Review.
Cited by
-
Insights from the molecular characterization of mercury stress proteins identified by proteomics in E.coli nissle 1917.Bioinformation. 2013 May 25;9(9):485-90. doi: 10.6026/97320630009485. Print 2013. Bioinformation. 2013. PMID: 23847405 Free PMC article.
-
New insight into poly (3-hydroxybutyrate) production by Azomonas macrocytogenes isolate KC685000: large scale production, kinetic modeling, recovery and characterization.Mol Biol Rep. 2019 Jun;46(3):3357-3370. doi: 10.1007/s11033-019-04798-4. Epub 2019 Apr 17. Mol Biol Rep. 2019. PMID: 30997598
-
PSSP-RFE: accurate prediction of protein structural class by recursive feature extraction from PSI-BLAST profile, physical-chemical property and functional annotations.PLoS One. 2014 Mar 27;9(3):e92863. doi: 10.1371/journal.pone.0092863. eCollection 2014. PLoS One. 2014. PMID: 24675610 Free PMC article.
-
Proposing a highly accurate protein structural class predictor using segmentation-based features.BMC Genomics. 2014;15 Suppl 1(Suppl 1):S2. doi: 10.1186/1471-2164-15-S1-S2. Epub 2014 Jan 24. BMC Genomics. 2014. PMID: 24564476 Free PMC article.
-
Many local pattern texture features: which is better for image-based multilabel human protein subcellular localization classification?ScientificWorldJournal. 2014;2014:429049. doi: 10.1155/2014/429049. Epub 2014 Jun 24. ScientificWorldJournal. 2014. PMID: 25050396 Free PMC article.
References
-
- Chou KC, Wei D, Du Q, Sirois S, Zhong W. Progress in computational approach to drug development against SARS. Curr Med Chem. 2006;13(32):63–70. - PubMed
-
- Chou KC. Structural bioinformatics and its impact to biomedical science. Curr Med Chem. 2004;11(21):05–34. - PubMed
-
- Kurgan LA, Cios KJ, Zhang H, Zhang T, Chen K, Shen S, Ruan J. Sequence-based methods for real value predictions of protein structure. Current Bioinformatics. 2008;3(3):183–196. doi: 10.2174/157489308785909197. - DOI
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
