

The other side of comparative genomics: genes with no orthologs between the cow and other mammalian species
- PMID: 20003425
- PMCID: PMC2808326
- DOI: 10.1186/1471-2164-10-604
The other side of comparative genomics: genes with no orthologs between the cow and other mammalian species
Abstract
Background: With the rapid growth in the availability of genome sequence data, the automated identification of orthologous genes between species (orthologs) is of fundamental importance to facilitate functional annotation and studies on comparative and evolutionary genomics. Genes with no apparent orthologs between the bovine and human genome may be responsible for major differences between the species, however, such genes are often neglected in functional genomics studies.
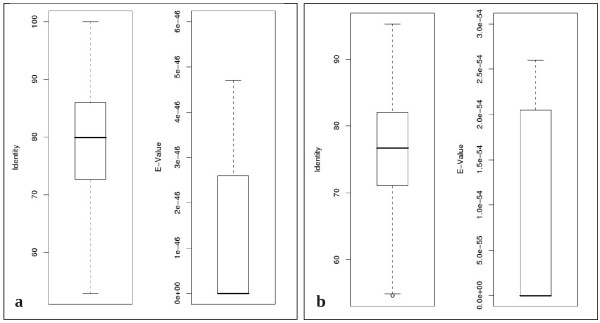
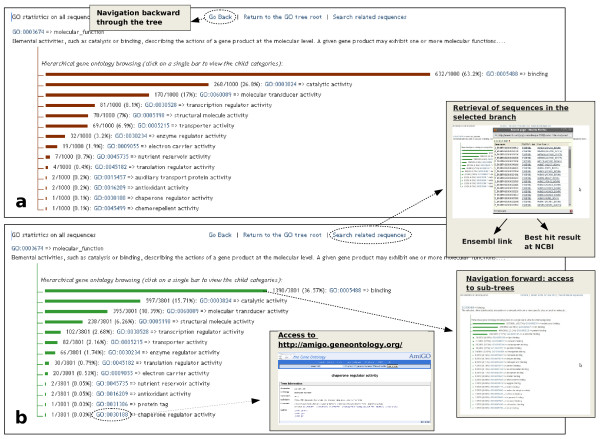
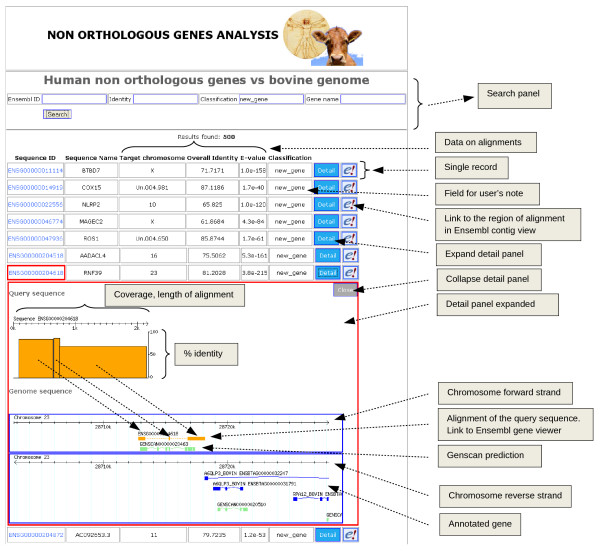
Results: A BLAST-based method was exploited to explore the current annotation and orthology predictions in Ensembl. Genes with no orthologs between the two genomes were classified into groups based on alignments, ontology, manual curation and publicly available information. Starting from a high quality and specific set of orthology predictions, as provided by Ensembl, hidden relationship between genes and genomes of different mammalian species were unveiled using a highly sensitive approach, based on sequence similarity and genomic comparison.
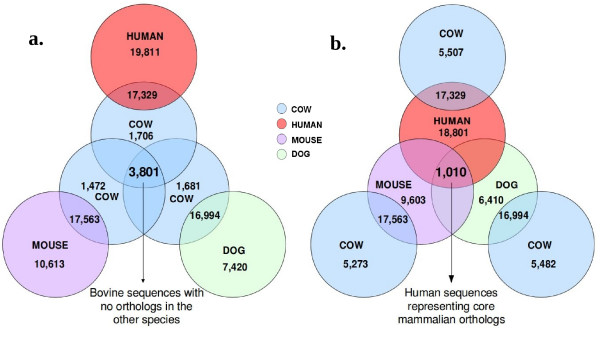
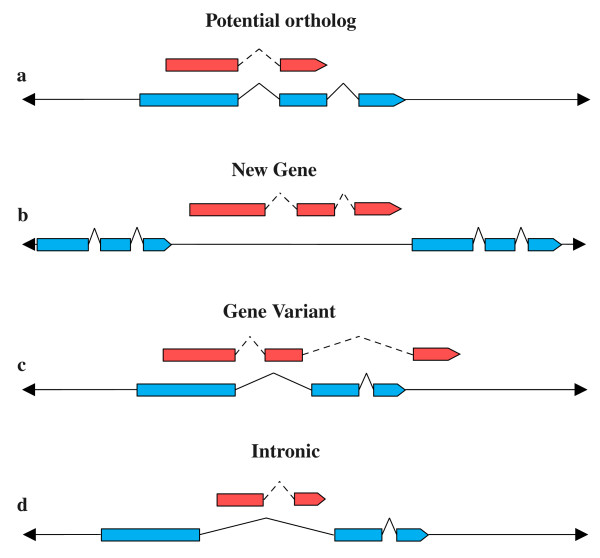
Conclusions: The analysis identified 3,801 bovine genes with no orthologs in human and 1010 human genes with no orthologs in cow, among which 411 and 43 genes, respectively, had no match at all in the other species. Most of the apparently non-orthologous genes may potentially have orthologs which were missed in the annotation process, despite having a high percentage of identity, because of differences in gene length and structure. The comparative analysis reported here identified gene variants, new genes and species-specific features and gave an overview of the other side of orthology which may help to improve the annotation of the bovine genome and the knowledge of structural differences between species.
Figures
Similar articles
-
Evola: Ortholog database of all human genes in H-InvDB with manual curation of phylogenetic trees.Nucleic Acids Res. 2008 Jan;36(Database issue):D787-92. doi: 10.1093/nar/gkm878. Epub 2007 Nov 3. Nucleic Acids Res. 2008. PMID: 17982176 Free PMC article.
-
Automatic clustering of orthologs and in-paralogs from pairwise species comparisons.J Mol Biol. 2001 Dec 14;314(5):1041-52. doi: 10.1006/jmbi.2000.5197. J Mol Biol. 2001. PMID: 11743721
-
Identification of mammalian orthologs using local synteny.BMC Genomics. 2009 Dec 23;10:630. doi: 10.1186/1471-2164-10-630. BMC Genomics. 2009. PMID: 20030836 Free PMC article.
-
Orthologs, paralogs, and evolutionary genomics.Annu Rev Genet. 2005;39:309-38. doi: 10.1146/annurev.genet.39.073003.114725. Annu Rev Genet. 2005. PMID: 16285863 Review.
-
[Reviews in comparative genomic research based on orthologs].Yi Chuan. 2009 May;31(5):457-63. doi: 10.3724/sp.j.1005.2009.00457. Yi Chuan. 2009. PMID: 19586838 Review. Chinese.
Cited by
-
Taxonomically restricted genes are associated with the evolution of sociality in the honey bee.BMC Genomics. 2011 Mar 29;12:164. doi: 10.1186/1471-2164-12-164. BMC Genomics. 2011. PMID: 21447185 Free PMC article.
-
Identification, characterization and expression analysis of lineage-specific genes within sweet orange (Citrus sinensis).BMC Genomics. 2015 Nov 23;16:995. doi: 10.1186/s12864-015-2211-z. BMC Genomics. 2015. PMID: 26597278 Free PMC article.
-
Genome-wide identification, characterization, and expression analysis of lineage-specific genes within zebrafish.BMC Genomics. 2013 Jan 31;14:65. doi: 10.1186/1471-2164-14-65. BMC Genomics. 2013. PMID: 23368736 Free PMC article.
-
Improved supervised prediction of aging-related genes via weighted dynamic network analysis.BMC Bioinformatics. 2021 Oct 25;22(1):520. doi: 10.1186/s12859-021-04439-3. BMC Bioinformatics. 2021. PMID: 34696741 Free PMC article.
-
MmCMS: mouse models' consensus molecular subtypes of colorectal cancer.Br J Cancer. 2023 Mar;128(7):1333-1343. doi: 10.1038/s41416-023-02157-6. Epub 2023 Jan 30. Br J Cancer. 2023. PMID: 36717674 Free PMC article.
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Research Materials
