

Large deletions and point mutations involving the dedicator of cytokinesis 8 (DOCK8) in the autosomal-recessive form of hyper-IgE syndrome
- PMID: 20004785
- PMCID: PMC2818862
- DOI: 10.1016/j.jaci.2009.10.038
Large deletions and point mutations involving the dedicator of cytokinesis 8 (DOCK8) in the autosomal-recessive form of hyper-IgE syndrome
Erratum in
- J Allergy Clin Immunol. 2010 Mar;125(3):743. Kutuculer, Necil [corrected to Kutukculer, Necil]
Abstract
Background: The genetic etiologies of the hyper-IgE syndromes are diverse. Approximately 60% to 70% of patients with hyper-IgE syndrome have dominant mutations in STAT3, and a single patient was reported to have a homozygous TYK2 mutation. In the remaining patients with hyper-IgE syndrome, the genetic etiology has not yet been identified.
Objectives: We aimed to identify a gene that is mutated or deleted in autosomal recessive hyper-IgE syndrome.
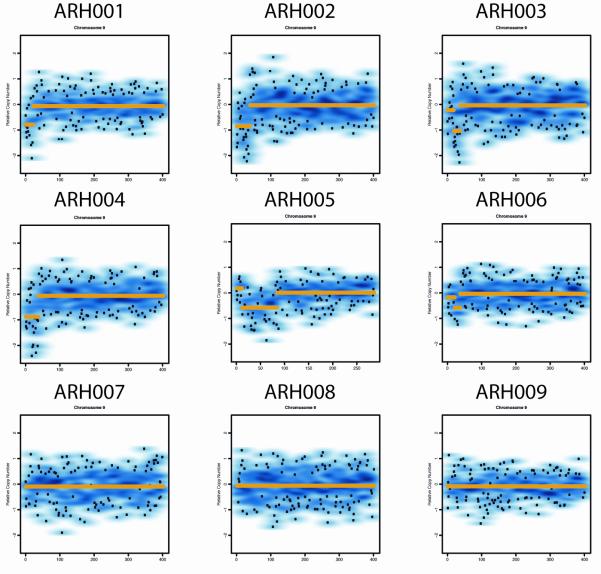
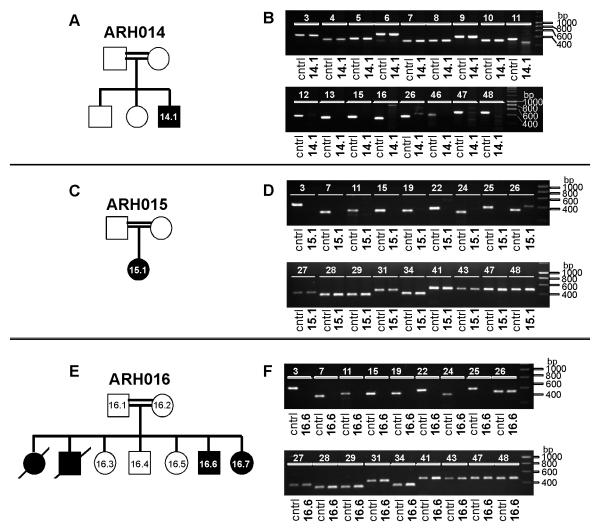
Methods: We performed genome-wide single nucleotide polymorphism analysis for 9 patients with autosomal-recessive hyper-IgE syndrome to locate copy number variations and homozygous haplotypes. Homozygosity mapping was performed with 12 patients from 7 additional families. The candidate gene was analyzed by genomic and cDNA sequencing to identify causative alleles in a total of 27 patients with autosomal-recessive hyper-IgE syndrome.
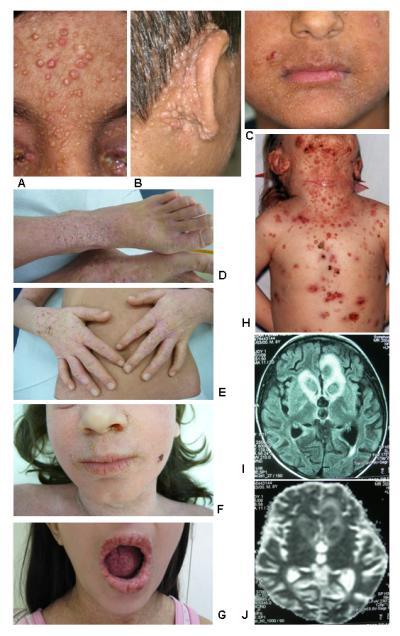
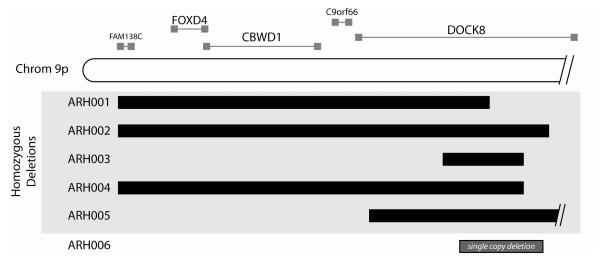
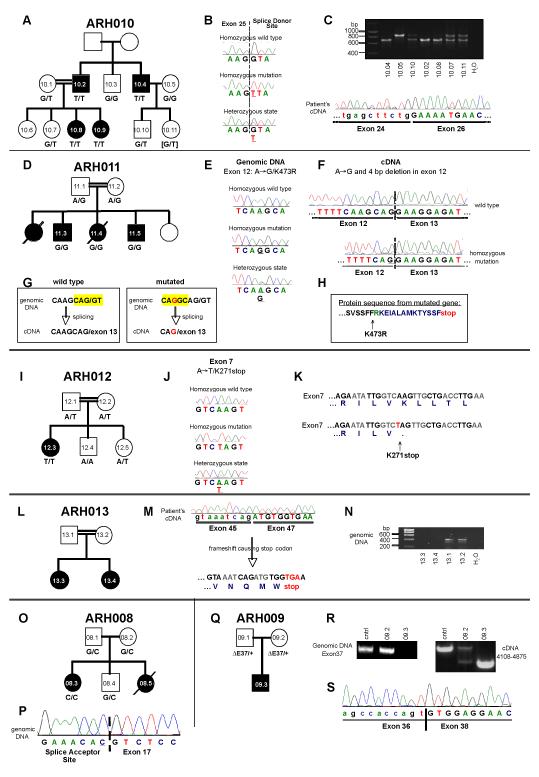
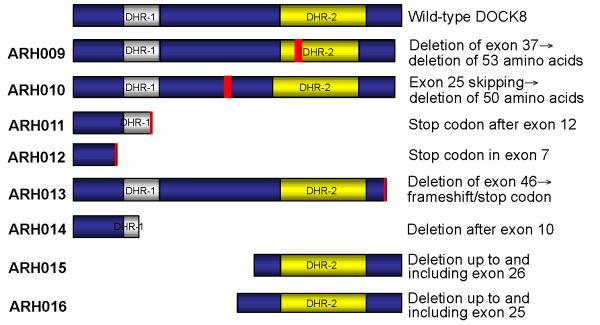
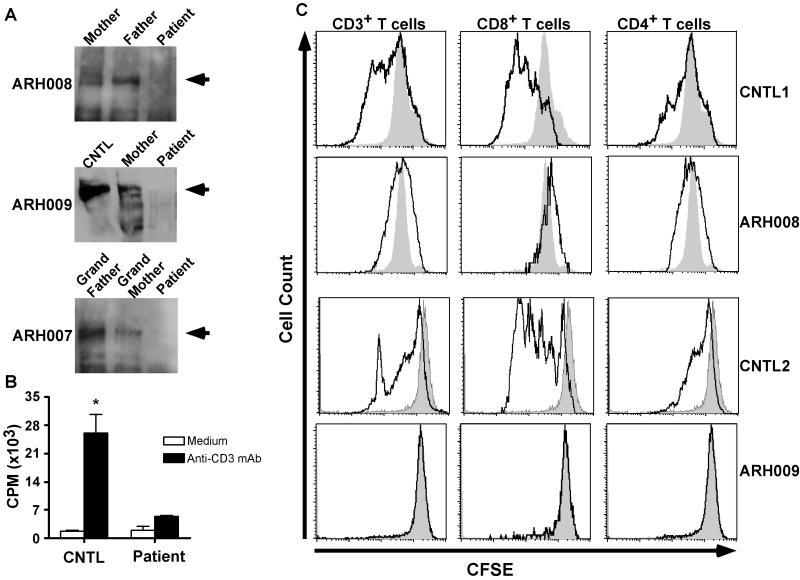
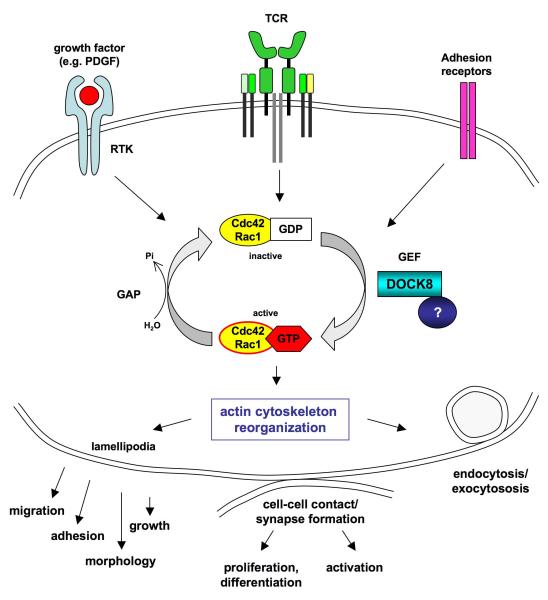
Results: Subtelomeric biallelic microdeletions were identified in 5 patients at the terminus of chromosome 9p. In all 5 patients, the deleted interval involved dedicator of cytokinesis 8 (DOCK8), encoding a protein implicated in the regulation of the actin cytoskeleton. Sequencing of patients without large deletions revealed 16 patients from 9 unrelated families with distinct homozygous mutations in DOCK8 causing premature termination, frameshift, splice site disruption, and single exon deletions and microdeletions. DOCK8 deficiency was associated with impaired activation of CD4+ and CD8+T cells.
Conclusion: Autosomal-recessive mutations in DOCK8 are responsible for many, although not all, cases of autosomal-recessive hyper-IgE syndrome. DOCK8 disruption is associated with a phenotype of severe cellular immunodeficiency characterized by susceptibility to viral infections, atopic eczema, defective T-cell activation and T(h)17 cell differentiation, and impaired eosinophil homeostasis and dysregulation of IgE.
Figures
References
-
- Grimbacher B, Holland SM, Gallin JI, Greenberg F, Hill SC, Malech HL, et al. Hyper-IgE syndrome with recurrent infections--an autosomal dominant multisystem disorder. N Engl J Med. 1999;340:692–702. - PubMed
-
- Grimbacher B, Holland SM, Puck JM. Hyper-IgE syndromes. Immunol Rev. 2005;203:244–50. - PubMed
-
- Holland SM, DeLeo FR, Elloumi HZ, Hsu AP, Uzel G, Brodsky N, et al. STAT3 mutations in the hyper-IgE syndrome. N Engl J Med. 2007;357:1608–19. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
