

Platelet polyphosphates are proinflammatory and procoagulant mediators in vivo
- PMID: 20005807
- PMCID: PMC2796262
- DOI: 10.1016/j.cell.2009.11.001
Platelet polyphosphates are proinflammatory and procoagulant mediators in vivo
Abstract
Platelets play a central role in thrombosis, hemostasis, and inflammation. We show that activated platelets release inorganic polyphosphate (polyP), a polymer of 60-100 phosphate residues that directly bound to and activated the plasma protease factor XII. PolyP-driven factor XII activation triggered release of the inflammatory mediator bradykinin by plasma kallikrein-mediated kininogen processing. PolyP increased vascular permeability and induced fluid extravasation in skin microvessels of mice. Mice deficient in factor XII or bradykinin receptors were resistant to polyP-induced leakage. PolyP initiated clotting of plasma via the contact pathway. Ablation of intrinsic coagulation pathway proteases factor XII and factor XI protected mice from polyP-triggered lethal pulmonary embolism. Targeting polyP with phosphatases interfered with procoagulant activity of activated platelets and blocked platelet-induced thrombosis in mice. Addition of polyP restored defective plasma clotting of Hermansky-Pudlak Syndrome patients, who lack platelet polyP. The data identify polyP as a new class of mediator having fundamental roles in platelet-driven proinflammatory and procoagulant disorders.
Figures
 ), 30 (
), 30 ( ), or 60 min (
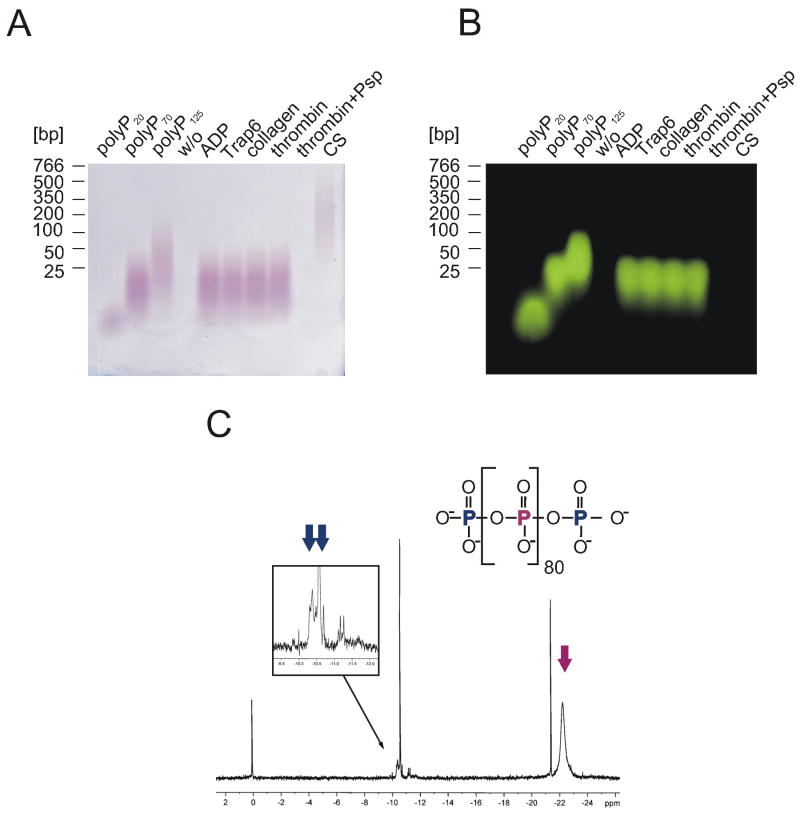
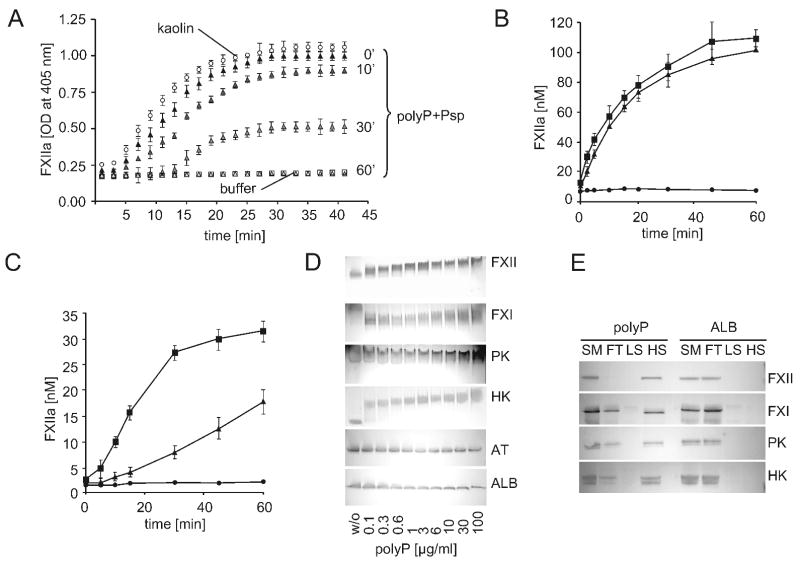
), or 60 min ( ). Data are means ± SD, n=5. (B and C) Plasma-free activation of purified FXII (200 nM) was analyzed in the presence (B) or absence (C) of PK (0.5 nM). The mixture was stimulated with dextran sulfate (■; 1 μg/ml), polyP (▲; 5 μg/ml), or buffer (●). FXIIa concentrations were determined using S-2302 hydrolysis. Data are means ± SD, n=4. (D) Gel-mobility shift analyses of polyP binding to proteins. Purified FXI, FXII, PK, HK, antithrombin (AT) or albumin (ALB) were incubated with buffer (“w/o”) or with increasing concentrations of polyP125 (0 - 100 μg) for 10 min, after which mixtures were resolved on a 10 % Tris-Glycine gel under native conditions and stained for protein using Gel Code Blue. (E) Protein binding to immobilized polyP. PolyP (left) or albumin (ALB, right) was coupled to zirconia (zirconium dioxide) beads. FXII, FXI, PK, or HK (5 μg each) were incubated with the beads, after which the flow-through (“FT”) fraction was collected by centrifugation using mini spin columns. The beads were washed with a low salt buffer (50 mM NaCl; “LS”) followed by a high salt buffer (1 M NaCl; “HS”). Fractions were probed by Western blotting with appropriate antibodies. Starting material (“SM”) refers to the initial protein load added to the beads.
). Data are means ± SD, n=5. (B and C) Plasma-free activation of purified FXII (200 nM) was analyzed in the presence (B) or absence (C) of PK (0.5 nM). The mixture was stimulated with dextran sulfate (■; 1 μg/ml), polyP (▲; 5 μg/ml), or buffer (●). FXIIa concentrations were determined using S-2302 hydrolysis. Data are means ± SD, n=4. (D) Gel-mobility shift analyses of polyP binding to proteins. Purified FXI, FXII, PK, HK, antithrombin (AT) or albumin (ALB) were incubated with buffer (“w/o”) or with increasing concentrations of polyP125 (0 - 100 μg) for 10 min, after which mixtures were resolved on a 10 % Tris-Glycine gel under native conditions and stained for protein using Gel Code Blue. (E) Protein binding to immobilized polyP. PolyP (left) or albumin (ALB, right) was coupled to zirconia (zirconium dioxide) beads. FXII, FXI, PK, or HK (5 μg each) were incubated with the beads, after which the flow-through (“FT”) fraction was collected by centrifugation using mini spin columns. The beads were washed with a low salt buffer (50 mM NaCl; “LS”) followed by a high salt buffer (1 M NaCl; “HS”). Fractions were probed by Western blotting with appropriate antibodies. Starting material (“SM”) refers to the initial protein load added to the beads.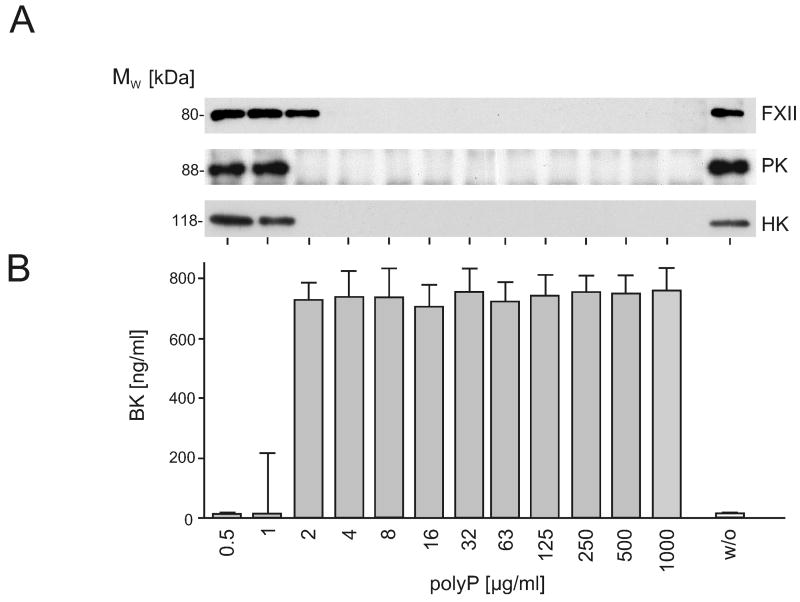
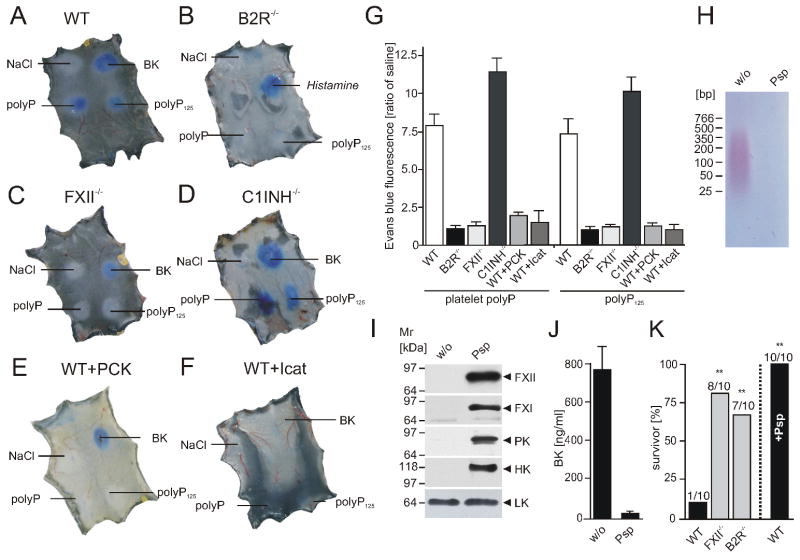
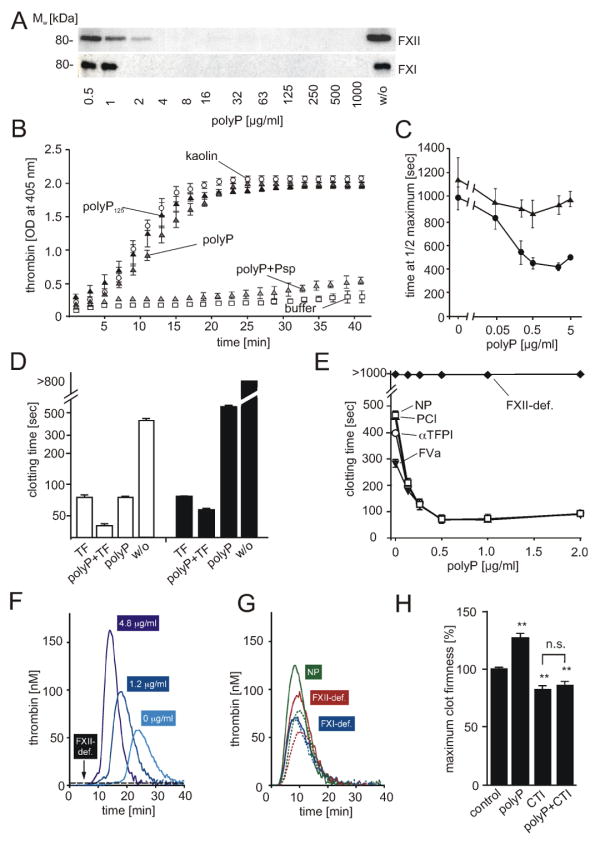 ; “polyP”), synthetic polyP125 (▲), platelet polyP treated with Psp prior to addition of plasma (; “polyP+Psp”). Data are means ± SD, n=5. (C) Plasma clotting times. PolyP was added to normal plasma (●) or FXII-immunodepleted plasma (▲) at a range of concentrations (0 - 5 μg/ml), with clot formation quantified as the half-maximal change in turbidity at 405 nm. Data are means ± SD, n=3. (D) Recalcification clotting times in normal (white columns) and FXII-deficient human plasma (black columns) incubated with TF (1 pM), polyP (3 μg/ml), TF and polyP (1 pM and 3 μg/ml), or buffer (w/o). Data are means ± SD, n=5. (E) Recalcification clotting times triggered by polyP (0 - 2 μg/ml) in FXII-deficient plasma (FXII-def.) and normal human plasma (NP) supplemented with FVa (4 nM, ▼), αTFPI antibody (10 μg/ml, □), or PCI (30 μg/ml, ○). Means ± SD, n=3. (F-H) Real time thrombin generation: (F) PolyP (0; 1.2; 4.8 μg/ml) triggered thrombin formation in plasma preincubated with an extrinsic pathway-inhibitor FVIIai. PolyP (4.8 μg/ml) failed to generate thrombin in FVIIai treated FXII-deficient plasma (FXII-def.). (G) Thrombin generation triggered by TF in FXII-deficient (red), FXI-deficient (blue), and normal plasma (green) in the absence (dashed lines) or presence of polyP (solid lines, 4.8 μg/ml). (H) Maximum clot firmness assessed by thromboelastography: Clot firmness in polyP (5 μg/ml), CTI (100 μg/ml), or polyP plus CTI-treated whole blood relative to untreated samples (100 %). Data are means ± SD, n=5.
; “polyP”), synthetic polyP125 (▲), platelet polyP treated with Psp prior to addition of plasma (; “polyP+Psp”). Data are means ± SD, n=5. (C) Plasma clotting times. PolyP was added to normal plasma (●) or FXII-immunodepleted plasma (▲) at a range of concentrations (0 - 5 μg/ml), with clot formation quantified as the half-maximal change in turbidity at 405 nm. Data are means ± SD, n=3. (D) Recalcification clotting times in normal (white columns) and FXII-deficient human plasma (black columns) incubated with TF (1 pM), polyP (3 μg/ml), TF and polyP (1 pM and 3 μg/ml), or buffer (w/o). Data are means ± SD, n=5. (E) Recalcification clotting times triggered by polyP (0 - 2 μg/ml) in FXII-deficient plasma (FXII-def.) and normal human plasma (NP) supplemented with FVa (4 nM, ▼), αTFPI antibody (10 μg/ml, □), or PCI (30 μg/ml, ○). Means ± SD, n=3. (F-H) Real time thrombin generation: (F) PolyP (0; 1.2; 4.8 μg/ml) triggered thrombin formation in plasma preincubated with an extrinsic pathway-inhibitor FVIIai. PolyP (4.8 μg/ml) failed to generate thrombin in FVIIai treated FXII-deficient plasma (FXII-def.). (G) Thrombin generation triggered by TF in FXII-deficient (red), FXI-deficient (blue), and normal plasma (green) in the absence (dashed lines) or presence of polyP (solid lines, 4.8 μg/ml). (H) Maximum clot firmness assessed by thromboelastography: Clot firmness in polyP (5 μg/ml), CTI (100 μg/ml), or polyP plus CTI-treated whole blood relative to untreated samples (100 %). Data are means ± SD, n=5.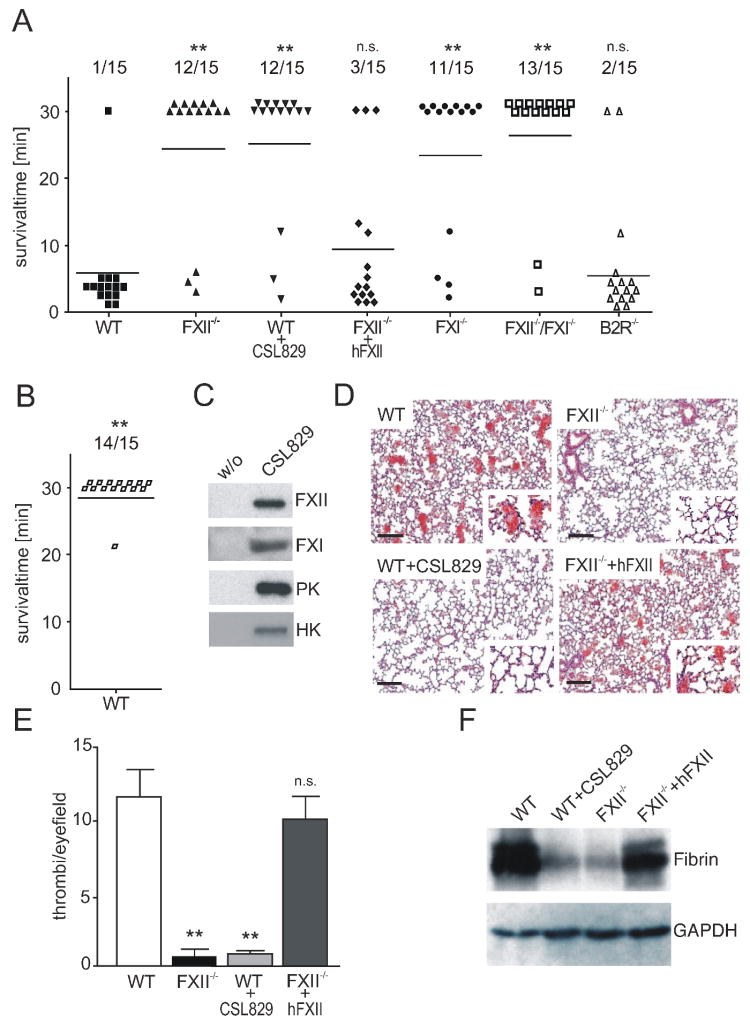
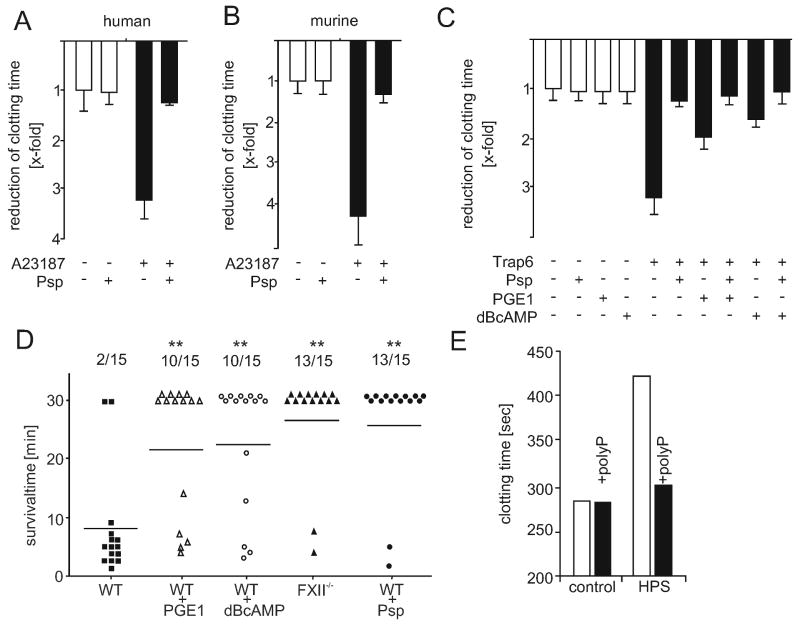
Comment in
-
Polyphosphates: a link between platelet activation, intrinsic coagulation and inflammation?Expert Rev Hematol. 2010 Jun;3(3):269-72. doi: 10.1586/ehm.10.26. Expert Rev Hematol. 2010. PMID: 21082980
References
-
- Castaldi PA, Larrieu MJ, Caen J. Availability of platelet Factor 3 and activation of factor XII in thrombasthenia. Nature. 1965;207:422–424. - PubMed
-
- Colman RW. In: Contact activation (Kallikrein-Kinin) Pathway: Multiple Physiologic and Pathophysiologic Activities. Colman RW, Mader VJ, Clowes AW, George JN, Goldhaber SZ, editors. Philadelphia: Lippincott Williams & Wilkins; 2006. pp. 107–130.
-
- Endler G, Marsik C, Jilma B, Schickbauer T, Quehenberger P, Mannhalter C. Evidence of a U-shaped association between factor XII activity and overall survival. J Thromb Haemost. 2007;5:1143–1148. - PubMed
-
- Furie B, Furie BC. Mechanisms of thrombus formation. N Engl J Med. 2008;359:938–949. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
