

A 2D mechanistic model of breast ductal carcinoma in situ (DCIS) morphology and progression
- PMID: 20006623
- PMCID: PMC2839055
- DOI: 10.1016/j.jtbi.2009.11.024
A 2D mechanistic model of breast ductal carcinoma in situ (DCIS) morphology and progression
Abstract
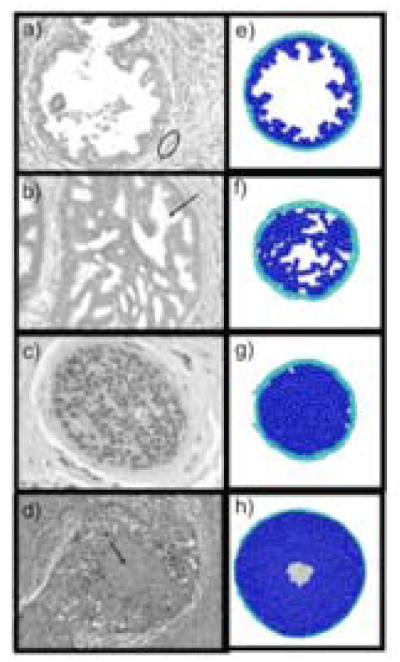
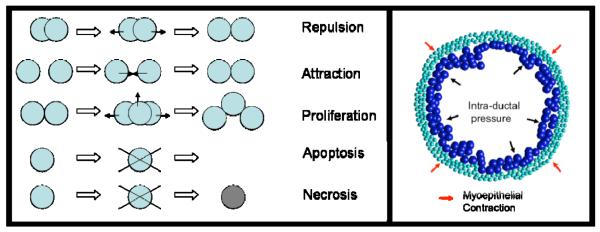
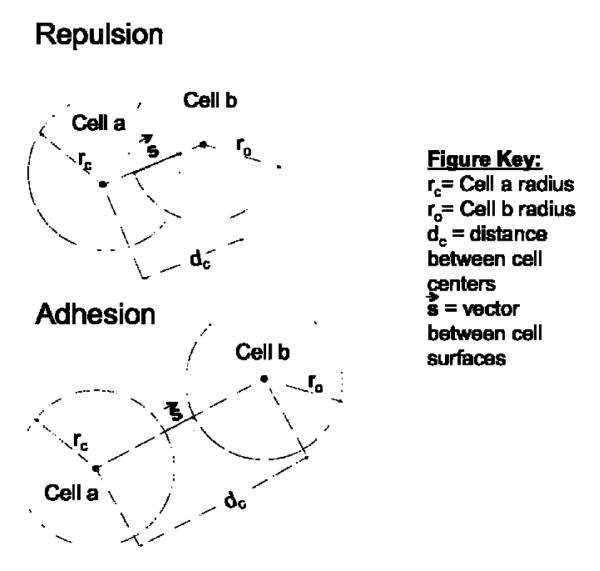
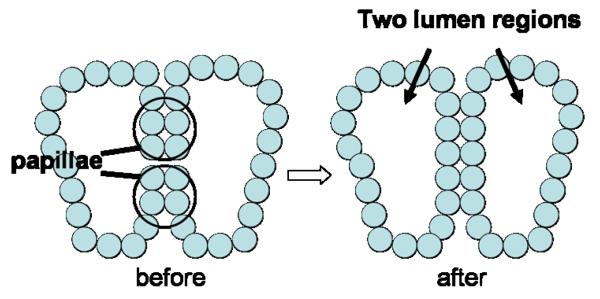
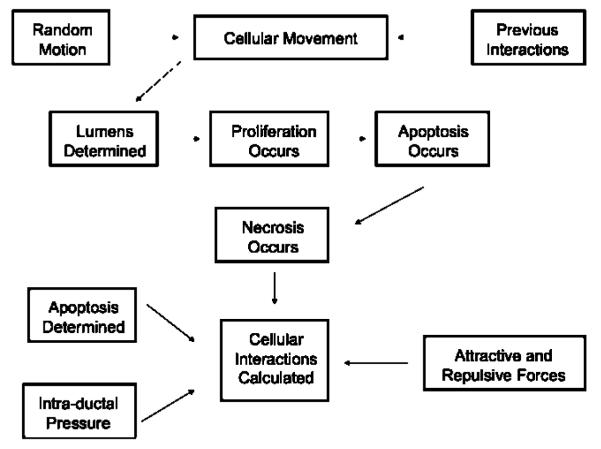
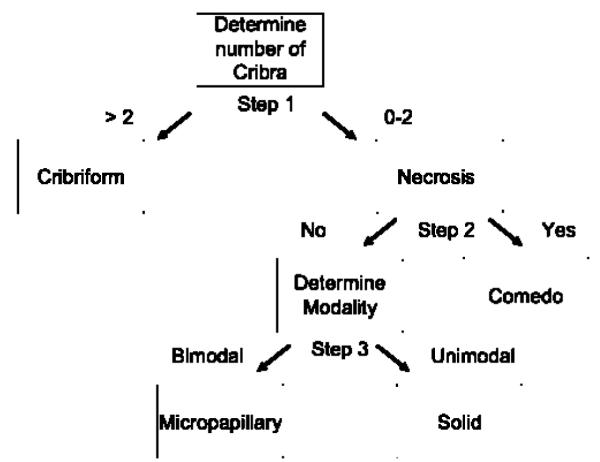
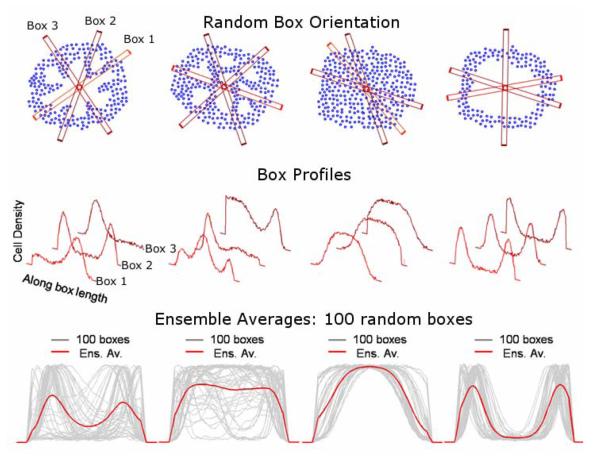
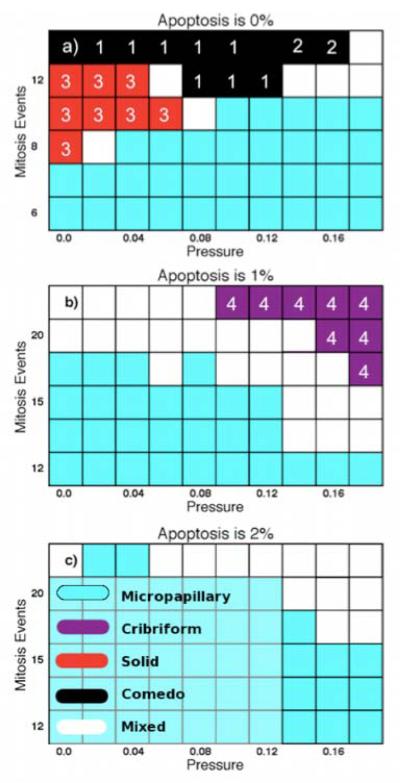
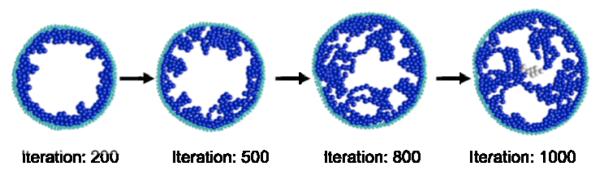
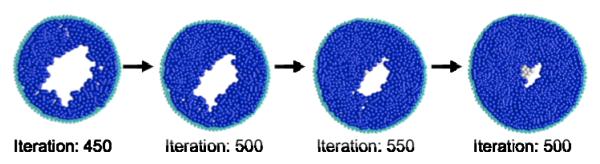
Ductal carcinoma in situ (DCIS) of the breast is a non-invasive tumor in which cells proliferate abnormally, but remain confined within a duct. Although four distinguishable DCIS morphologies are recognized, the mechanisms that generate these different morphological classes remain unclear, and consequently the prognostic strength of DCIS classification is not strong. To improve the understanding of the relation between morphology and time course, we have developed a 2D in silico particle model of the growth of DCIS within a single breast duct. This model considers mechanical effects such as cellular adhesion and intra-ductal pressure, and biological features including proliferation, apoptosis, necrosis, and cell polarity. Using this model, we find that different regions of parameter space generate distinct morphological subtypes of DCIS, so elucidating the relation between morphology and time course. Furthermore, we find that tumors with similar architectures may in fact be produced through different mechanisms, and we propose future work to further disentangle the mechanisms involved in DCIS progression.
(c) 2009 Elsevier Ltd. All rights reserved.
Figures

Similar articles
-
Evidence that molecular changes in cells occur before morphological alterations during the progression of breast ductal carcinoma.Breast Cancer Res. 2008;10(5):R87. doi: 10.1186/bcr2157. Epub 2008 Oct 17. Breast Cancer Res. 2008. PMID: 18928525 Free PMC article.
-
Silencing of HSulf-2 expression in MCF10DCIS.com cells attenuate ductal carcinoma in situ progression to invasive ductal carcinoma in vivo.Breast Cancer Res. 2012 Mar 12;14(2):R43. doi: 10.1186/bcr3140. Breast Cancer Res. 2012. PMID: 22410125 Free PMC article.
-
Lattice-based model of ductal carcinoma in situ suggests rules for breast cancer progression to an invasive state.PLoS Comput Biol. 2014 Dec 4;10(12):e1003997. doi: 10.1371/journal.pcbi.1003997. eCollection 2014 Dec. PLoS Comput Biol. 2014. PMID: 25473842 Free PMC article.
-
The molecular journey from ductal carcinoma in situ to invasive breast cancer.Cancer. 2008 May 15;112(10):2130-42. doi: 10.1002/cncr.23430. Cancer. 2008. PMID: 18383519 Review.
-
Models for Studying Ductal Carcinoma In Situ Progression.Adv Exp Med Biol. 2025;1464:95-108. doi: 10.1007/978-3-031-70875-6_6. Adv Exp Med Biol. 2025. PMID: 39821022 Review.
Cited by
-
Phenotypic transition maps of 3D breast acini obtained by imaging-guided agent-based modeling.Integr Biol (Camb). 2011 Apr;3(4):408-21. doi: 10.1039/c0ib00092b. Epub 2011 Mar 4. Integr Biol (Camb). 2011. PMID: 21373705 Free PMC article.
-
Mathematical Oncology: How Are the Mathematical and Physical Sciences Contributing to the War on Breast Cancer?Curr Breast Cancer Rep. 2010 Sep;2(3):121-129. doi: 10.1007/s12609-010-0020-6. Epub 2010 Jul 22. Curr Breast Cancer Rep. 2010. PMID: 21151486 Free PMC article.
-
Tumor growth and calcification in evolving microenvironmental geometries.J Theor Biol. 2019 Feb 21;463:138-154. doi: 10.1016/j.jtbi.2018.12.006. Epub 2018 Dec 5. J Theor Biol. 2019. PMID: 30528340 Free PMC article.
-
Towards personalized computational oncology: from spatial models of tumour spheroids, to organoids, to tissues.J R Soc Interface. 2018 Jan;15(138):20170703. doi: 10.1098/rsif.2017.0703. J R Soc Interface. 2018. PMID: 29367239 Free PMC article. Review.
-
An agent-based model of cancer stem cell initiated avascular tumour growth and metastasis: the effect of seeding frequency and location.J R Soc Interface. 2014 Nov 6;11(100):20140640. doi: 10.1098/rsif.2014.0640. J R Soc Interface. 2014. PMID: 25185580 Free PMC article.
References
-
- Alarcón T, Byrne H, Maini P. A cellular automaton model for tumour growth in an inhomogeneous environment. Journal of Theoretical Biology. 2003;225:257–274. - PubMed
-
- Allred DC, Wu Y, Mao S, Nagtegaal ID, Lee S, Perou CM, Mohsin SK, O’Connell P, Tsimelzon A, Medina D. Ductal carcinoma in situ and the emergence of diversity during breast cancer evolution. Clin Cancer Res. 2008;14:370–8. - PubMed
-
- Ambrosi D, Mollica F. On the mechanics of a growing tumor. Int J Eng Sci. 2002;40:1297–1316.
-
- Anderson A, Weaver A, Cummings P, Quaranta V. Tumor morphology and phenotypic evolution driven by selective pressure from the microenvironment. Cell. 2006;127:905–915. - PubMed
-
- Anderson AR, Quaranta V. Integrative mathematical oncology. Nat Rev Cancer. 2008;8:227–34. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
