

Hepatitis C virus blocks interferon effector function by inducing protein kinase R phosphorylation
- PMID: 20006840
- PMCID: PMC2905238
- DOI: 10.1016/j.chom.2009.11.004
Hepatitis C virus blocks interferon effector function by inducing protein kinase R phosphorylation
Abstract
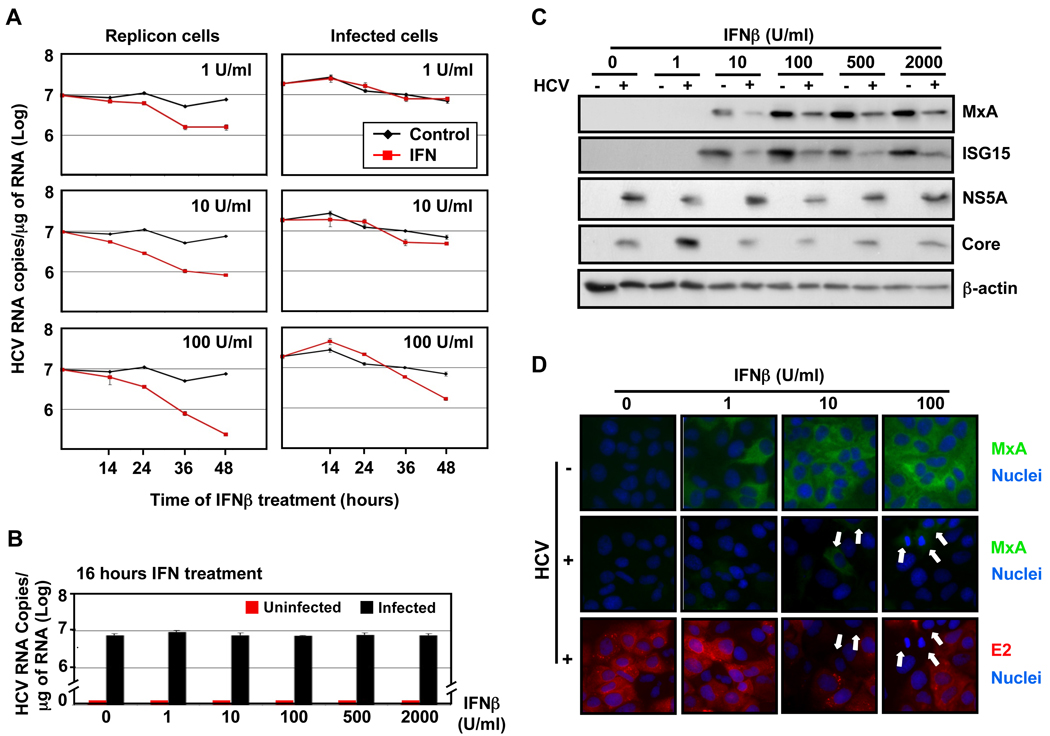
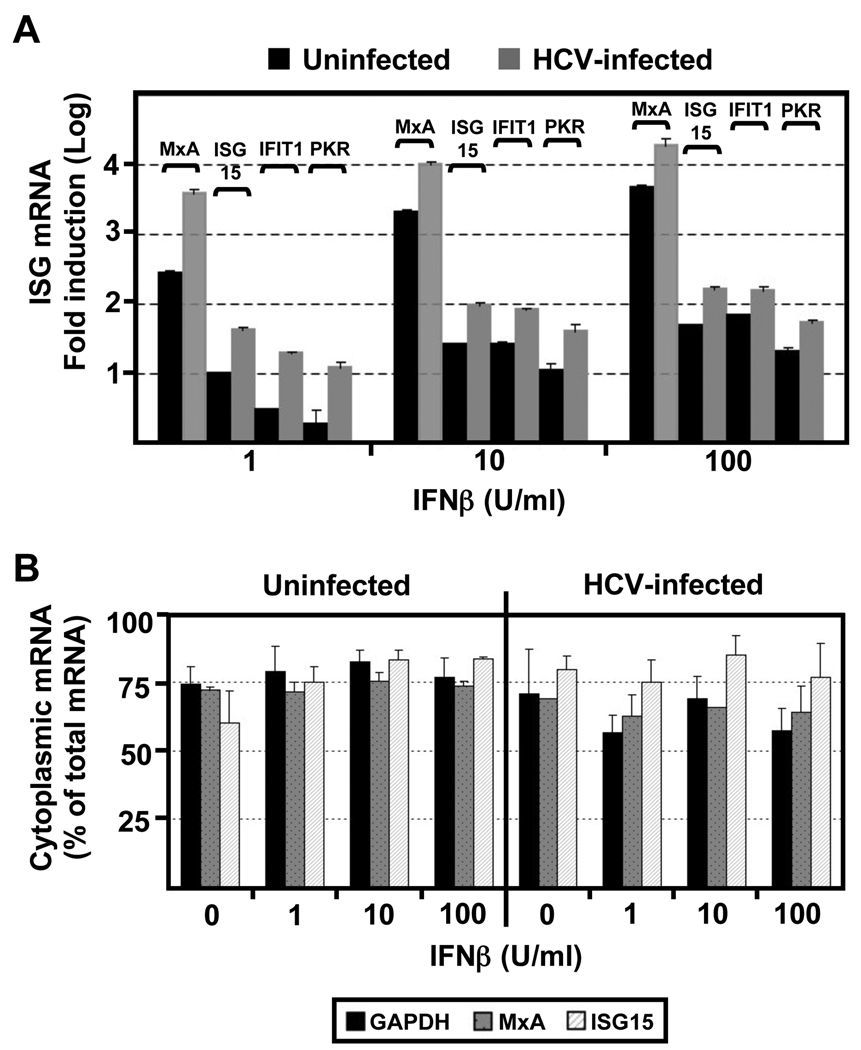
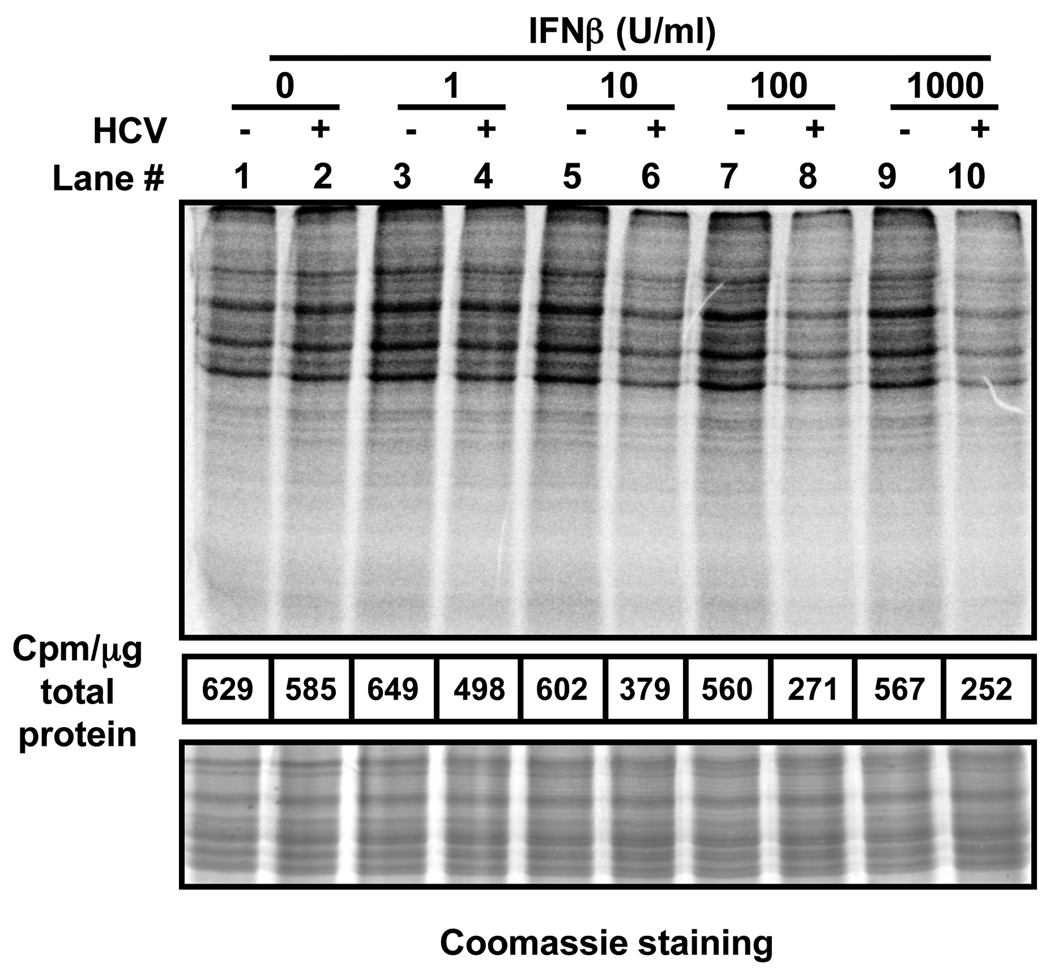
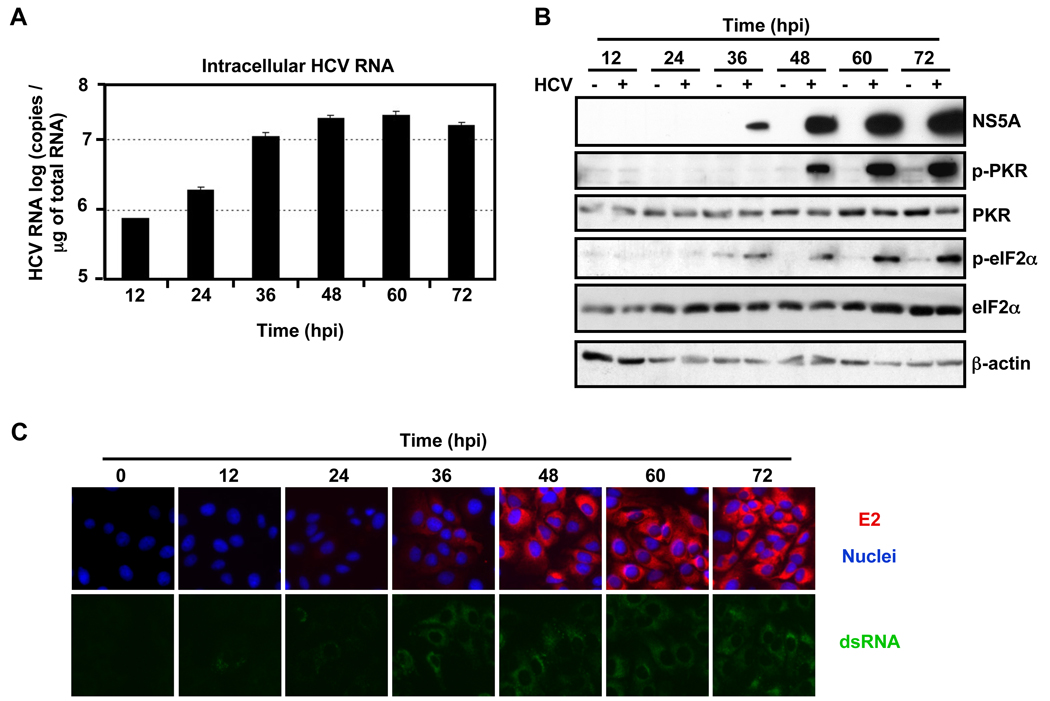
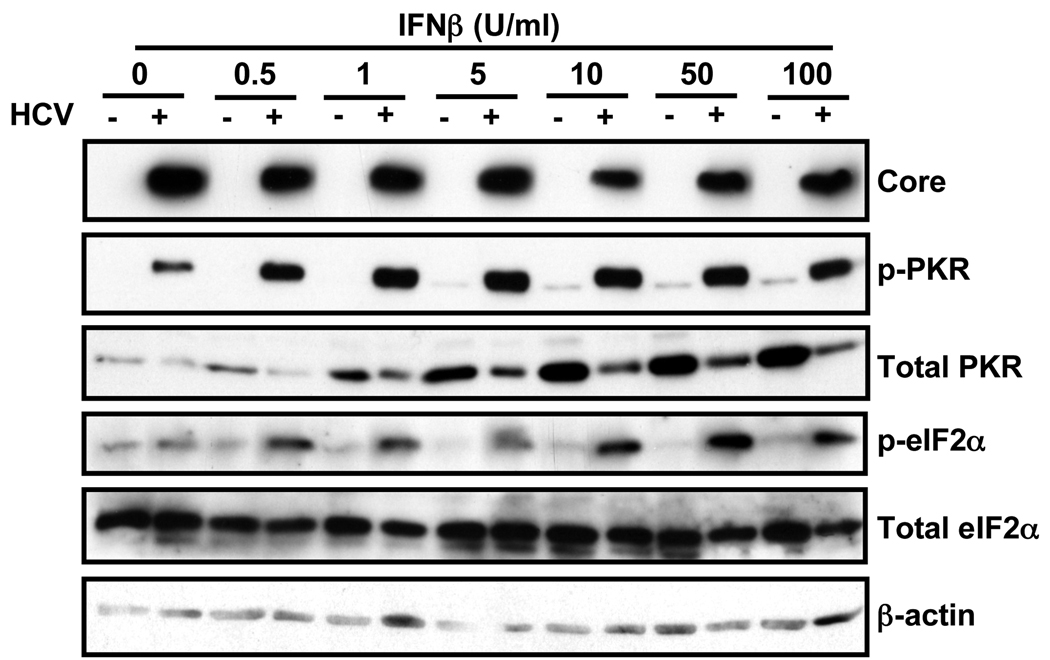
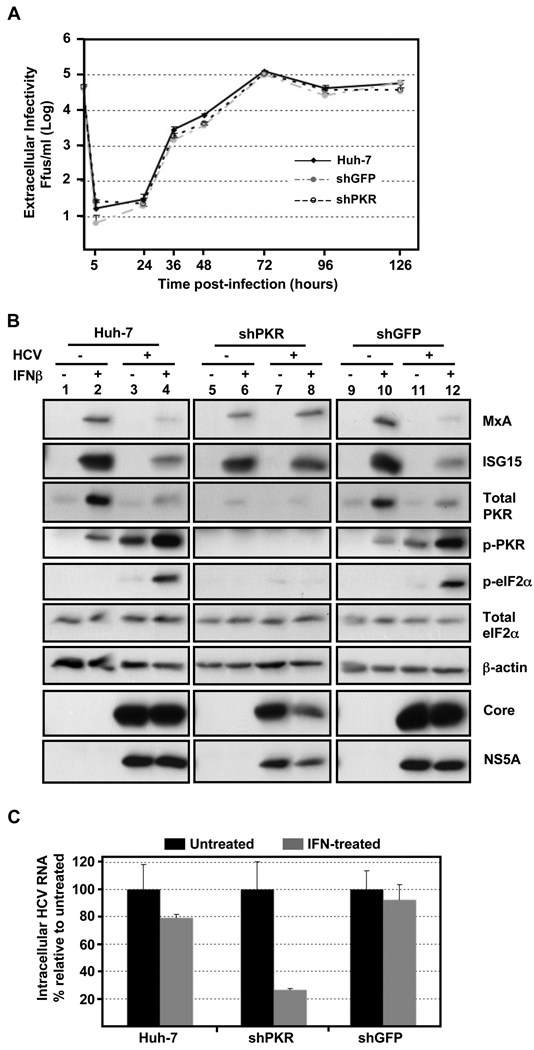
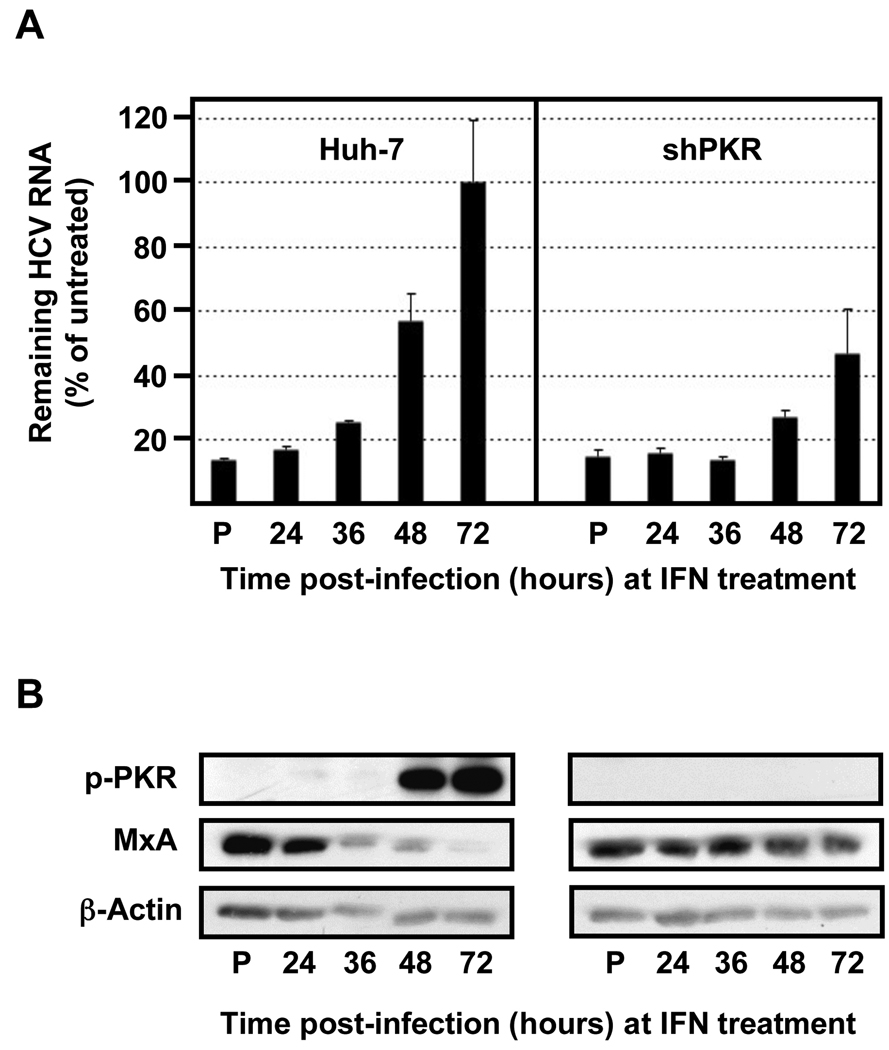
Hepatitis C virus (HCV) is a single-stranded RNA virus encoding a single polyprotein whose translation is driven by an internal ribosome entry site (IRES). HCV infection strongly induces antiviral interferon-stimulated gene (ISG) expression in the liver, yet it persists, suggesting that HCV can block ISG effector function. We now show that HCV infection triggers phosphorylation and activation of the RNA-dependent protein kinase PKR, which inhibits eukaryotic translation initiation factor eIF2 alpha and attenuates ISG protein expression despite normal ISG mRNA induction. ISG protein induction is restored and the antiviral effects of interferon are enhanced when PKR expression is suppressed in interferon-treated infected cells. Whereas host protein translation, including antiviral ISGs, is suppressed by activated PKR, HCV IRES-dependent translation is not. These results suggest that the ability of HCV to activate PKR may, paradoxically, be advantageous for the virus during an IFN response by preferentially suppressing the translation of ISGs.
Figures
Comment in
-
Hepatitis C and evasion of the interferon system: a PKR paradigm.Cell Host Microbe. 2009 Dec 17;6(6):495-7. doi: 10.1016/j.chom.2009.11.009. Cell Host Microbe. 2009. PMID: 20006836
References
-
- Alter HJ, Seeff LB. Recovery, persistence, and sequelae in hepatitis C virus infection: a perspective on long-term outcome. Semin Liver Dis. 2000;20:17–35. - PubMed
-
- Deutsch M, Hadziyannis SJ. Old and emerging therapies in chronic hepatitis C: an update. J Viral Hepat. 2008;15:2–11. - PubMed
-
- Dreux M, Boson B, Ricard-Blum S, Molle J, Lavillette D, Bartosch B, Pecheur EI, Cosset FL. The exchangeable apolipoprotein ApoC-I promotes membrane fusion of hepatitis C virus. J Biol Chem. 2007;282:32357–32369. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
