

Negative autoregulation of GTF2IRD1 in Williams-Beuren syndrome via a novel DNA binding mechanism
- PMID: 20007321
- PMCID: PMC2836076
- DOI: 10.1074/jbc.M109.086660
Negative autoregulation of GTF2IRD1 in Williams-Beuren syndrome via a novel DNA binding mechanism
Abstract
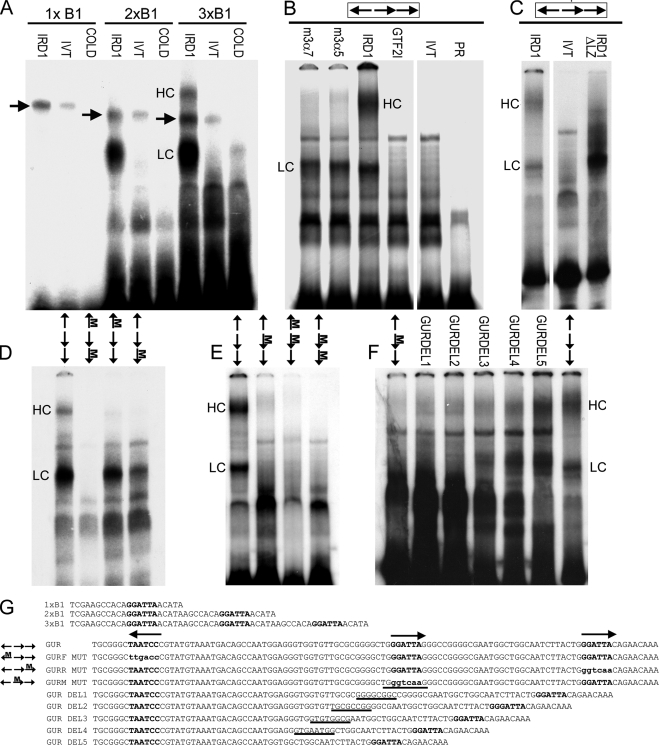
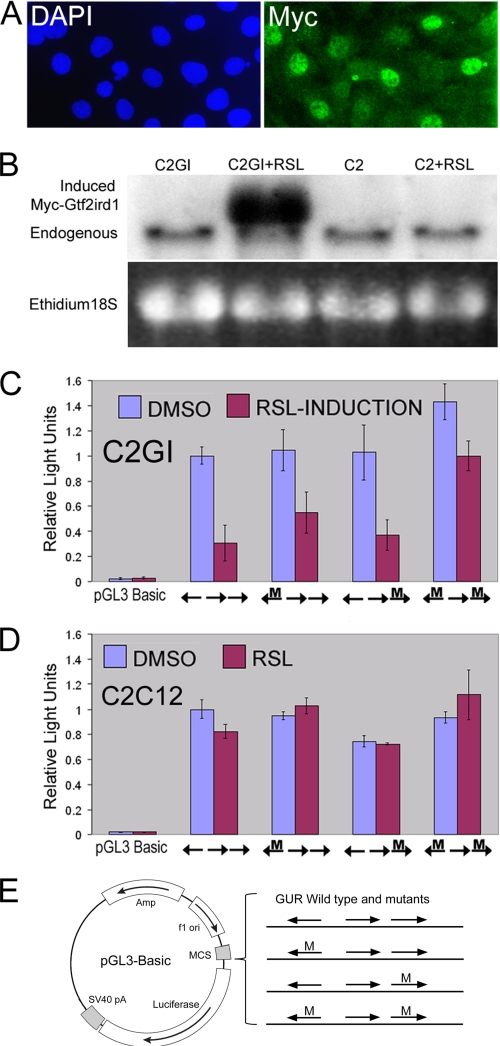
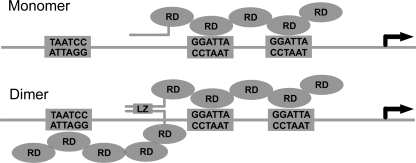
The GTF2IRD1 gene is of principal interest to the study of Williams-Beuren syndrome (WBS). This neurodevelopmental disorder results from the hemizygous deletion of a region of chromosome 7q11.23 containing 28 genes including GTF2IRD1. WBS is thought to be caused by haploinsufficiency of certain dosage-sensitive genes within the deleted region, and the feature of supravalvular aortic stenosis (SVAS) has been attributed to reduced elastin caused by deletion of ELN. Human genetic mapping data have implicated two related genes GTF2IRD1 and GTF2I in the cause of some the key features of WBS, including craniofacial dysmorphology, hypersociability, and visuospatial deficits. Mice with mutations of the Gtf2ird1 allele show evidence of craniofacial abnormalities and behavioral changes. Here we show the existence of a negative autoregulatory mechanism that controls the level of GTF2IRD1 transcription via direct binding of the GTF2IRD1 protein to a highly conserved region of the GTF2IRD1 promoter containing an array of three binding sites. The affinity for this protein-DNA interaction is critically dependent upon multiple interactions between separate domains of the protein and at least two of the DNA binding sites. This autoregulatory mechanism leads to dosage compensation of GTF2IRD1 transcription in WBS patients. The GTF2IRD1 promoter represents the first established in vivo gene target of the GTF2IRD1 protein, and we use it to model its DNA interaction capabilities.
Figures
Similar articles
-
RNA-Seq analysis of Gtf2ird1 knockout epidermal tissue provides potential insights into molecular mechanisms underpinning Williams-Beuren syndrome.BMC Genomics. 2016 Jun 13;17:450. doi: 10.1186/s12864-016-2801-4. BMC Genomics. 2016. PMID: 27295951 Free PMC article.
-
Partial 7q11.23 deletions further implicate GTF2I and GTF2IRD1 as the main genes responsible for the Williams-Beuren syndrome neurocognitive profile.J Med Genet. 2010 May;47(5):312-20. doi: 10.1136/jmg.2009.071712. Epub 2009 Nov 5. J Med Genet. 2010. PMID: 19897463
-
Global analysis of gene expression in the developing brain of Gtf2ird1 knockout mice.PLoS One. 2011;6(8):e23868. doi: 10.1371/journal.pone.0023868. Epub 2011 Aug 31. PLoS One. 2011. PMID: 21909369 Free PMC article.
-
[Williams-Beuren syndrome: a multidisciplinary approach].Arch Pediatr. 2009 Mar;16(3):273-82. doi: 10.1016/j.arcped.2008.11.011. Epub 2008 Dec 18. Arch Pediatr. 2009. PMID: 19097873 Review. French.
-
Genetic aspects of supravalvular aortic stenosis.Curr Opin Cardiol. 1998 May;13(3):214-9. Curr Opin Cardiol. 1998. PMID: 9649945 Review.
Cited by
-
The role of GTF2IRD1 in the auditory pathology of Williams-Beuren Syndrome.Eur J Hum Genet. 2015 Jun;23(6):774-80. doi: 10.1038/ejhg.2014.188. Epub 2014 Sep 24. Eur J Hum Genet. 2015. PMID: 25248400 Free PMC article.
-
Williams Syndrome, Human Self-Domestication, and Language Evolution.Front Psychol. 2019 Mar 18;10:521. doi: 10.3389/fpsyg.2019.00521. eCollection 2019. Front Psychol. 2019. PMID: 30936846 Free PMC article.
-
Copy number variants at Williams-Beuren syndrome 7q11.23 region.Hum Genet. 2010 Jul;128(1):3-26. doi: 10.1007/s00439-010-0827-2. Epub 2010 May 1. Hum Genet. 2010. PMID: 20437059 Review.
-
Functions of Gtf2i and Gtf2ird1 in the developing brain: transcription, DNA binding and long-term behavioral consequences.Hum Mol Genet. 2020 Jun 3;29(9):1498-1519. doi: 10.1093/hmg/ddaa070. Hum Mol Genet. 2020. PMID: 32313931 Free PMC article.
-
Animal models of Williams syndrome.Am J Med Genet C Semin Med Genet. 2010 May 15;154C(2):209-19. doi: 10.1002/ajmg.c.30257. Am J Med Genet C Semin Med Genet. 2010. PMID: 20425782 Free PMC article. Review.
References
-
- Strømme P., Bjørnstad P. G., Ramstad K. (2002) J. Child Neurol. 17, 269–271 - PubMed
-
- Valero M. C., de Luis O., Cruces J., Pérez Jurado L. A. (2000) Genomics 69, 1–13 - PubMed
-
- Curran M. E., Atkinson D. L., Ewart A. K., Morris C. A., Leppert M. F., Keating M. T. (1993) Cell 73, 159–168 - PubMed
-
- Hirota H., Matsuoka R., Chen X. N., Salandanan L. S., Lincoln A., Rose F. E., Sunahara M., Osawa M., Bellugi U., Korenberg J. R. (2003) Genet. Med. 5, 311–321 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
