

Insulin gene mutations resulting in early-onset diabetes: marked differences in clinical presentation, metabolic status, and pathogenic effect through endoplasmic reticulum retention
- PMID: 20007936
- PMCID: PMC2828668
- DOI: 10.2337/db09-1091
Insulin gene mutations resulting in early-onset diabetes: marked differences in clinical presentation, metabolic status, and pathogenic effect through endoplasmic reticulum retention
Abstract
Objective: Heterozygous mutations in the human preproinsulin (INS) gene are a cause of nonsyndromic neonatal or early-infancy diabetes. Here, we sought to identify INS mutations associated with maturity-onset diabetes of the young (MODY) or nonautoimmune diabetes in mid-adult life, and to explore the molecular mechanisms involved.
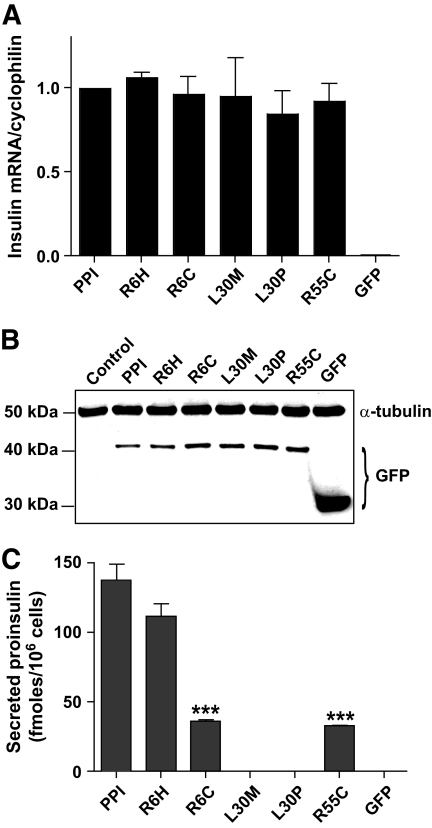
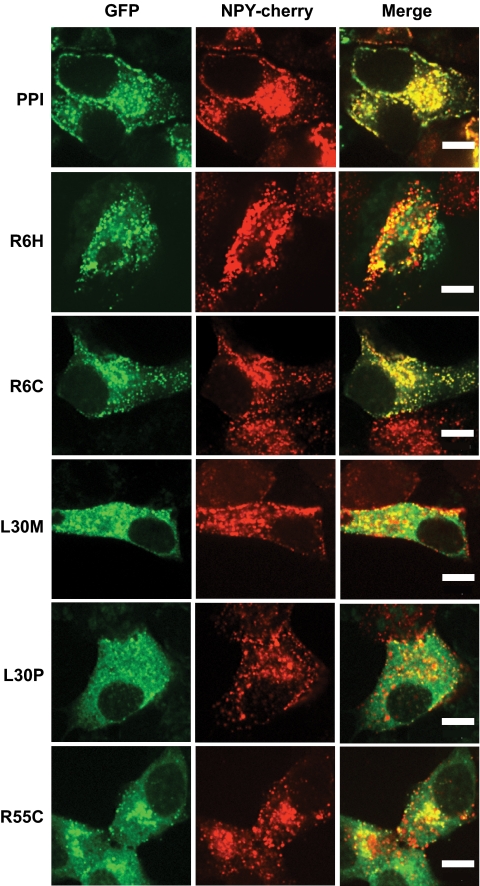
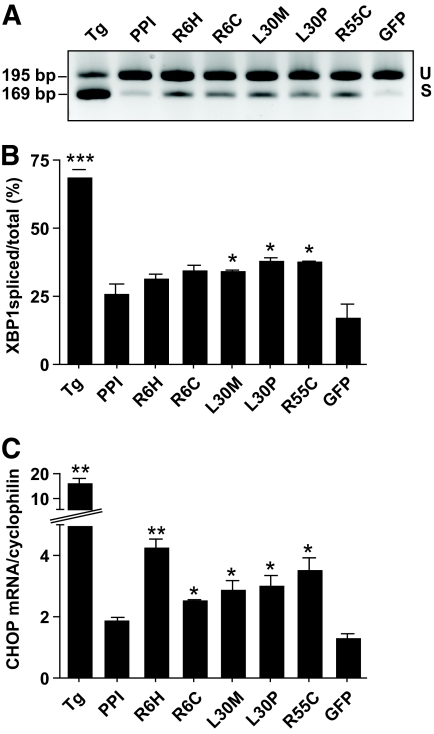
Research design and methods: The INS gene was sequenced in 16 French probands with unexplained MODY, 95 patients with nonautoimmune early-onset diabetes (diagnosed at <35 years) and 292 normoglycemic control subjects of French origin. Three identified insulin mutants were generated by site-directed mutagenesis of cDNA encoding a preproinsulin-green fluorescent protein (GFP) (C-peptide) chimera. Intracellular targeting was assessed in clonal beta-cells by immunocytochemistry and proinsulin secretion, by radioimmunoassay. Spliced XBP1 and C/EBP homologous protein were quantitated by real-time PCR.
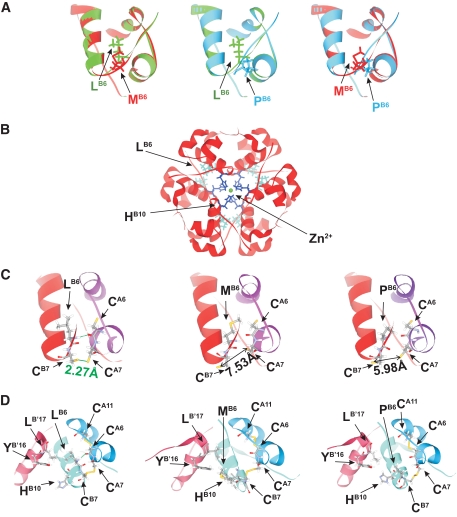
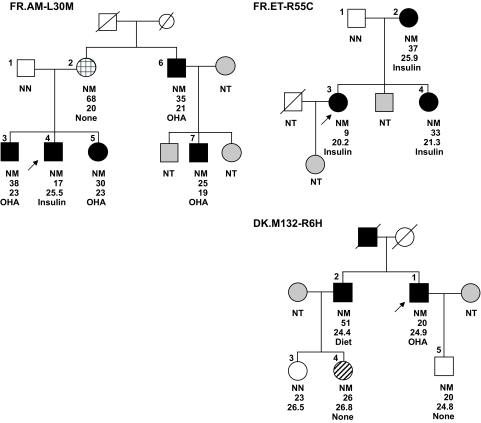
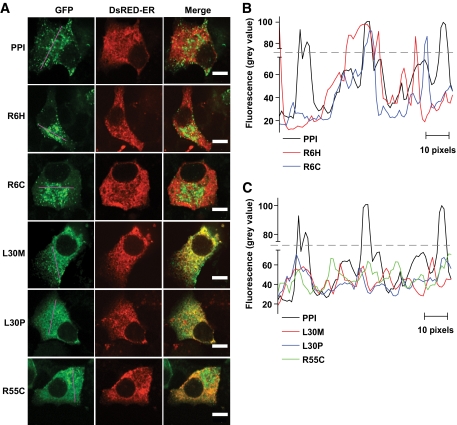
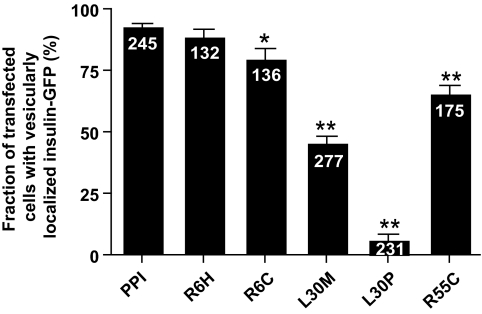
Results: A novel coding mutation, L30M, potentially affecting insulin multimerization, was identified in five diabetic individuals (diabetes onset 17-36 years) in a single family. L30M preproinsulin-GFP fluorescence largely associated with the endoplasmic reticulum (ER) in MIN6 beta-cells, and ER exit was inhibited by approximately 50%. Two additional mutants, R55C (at the B/C junction) and R6H (in the signal peptide), were normally targeted to secretory granules, but nonetheless caused substantial ER stress.
Conclusions: We describe three INS mutations cosegregating with early-onset diabetes whose clinical presentation is compatible with MODY. These led to the production of (pre)proinsulin molecules with markedly different trafficking properties and effects on ER stress, demonstrating a range of molecular defects in the beta-cell.
Figures
References
-
- Herbach N, Rathkolb B, Kemter E, Pichl L, Klaften M, de Angelis MH, Halban PA, Wolf E, Aigner B, Wanke R: Dominant-negative effects of a novel mutated Ins2 allele causes early-onset diabetes and severe beta-cell loss in Munich Ins2C95S mutant mice. Diabetes 2007; 56: 1268– 1276 - PubMed
-
- Shoelson S, Haneda M, Blix P, Nanjo A, Sanke T, Inouye K, Steiner D, Rubenstein A, Tager H: Three mutant insulins in man. Nature 1983; 302: 540– 543 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
