

Prdm16 is required for normal palatogenesis in mice
- PMID: 20007998
- PMCID: PMC2816611
- DOI: 10.1093/hmg/ddp543
Prdm16 is required for normal palatogenesis in mice
Abstract
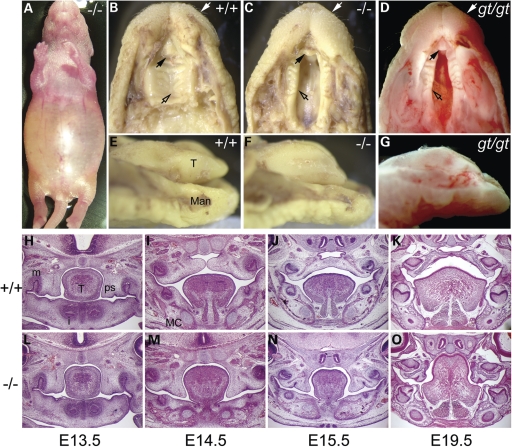
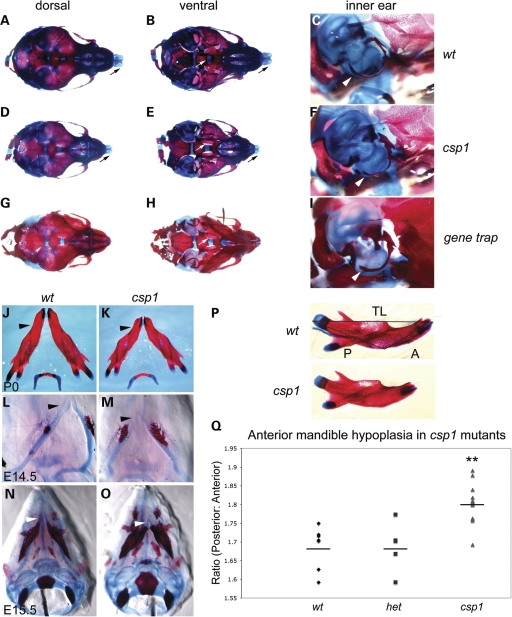
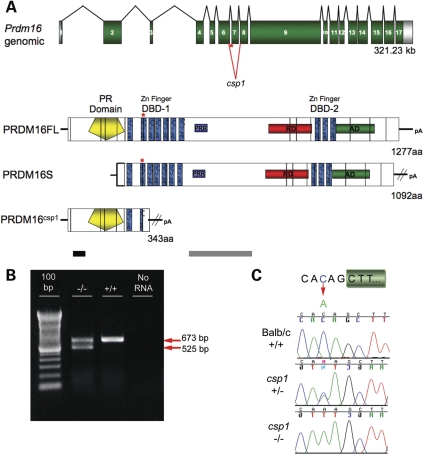
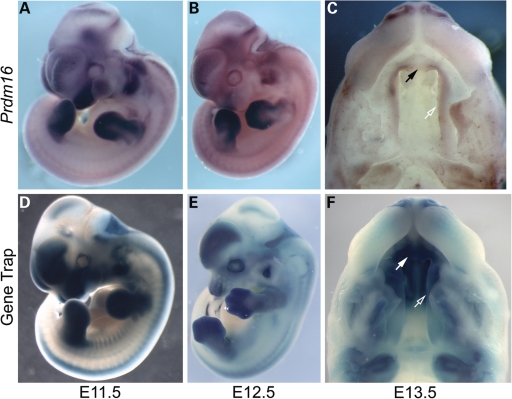
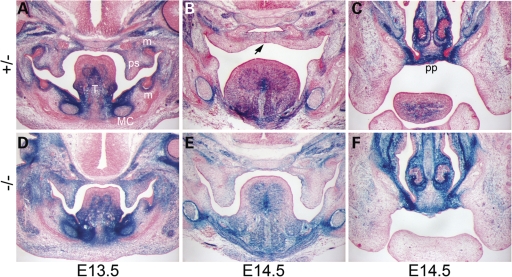
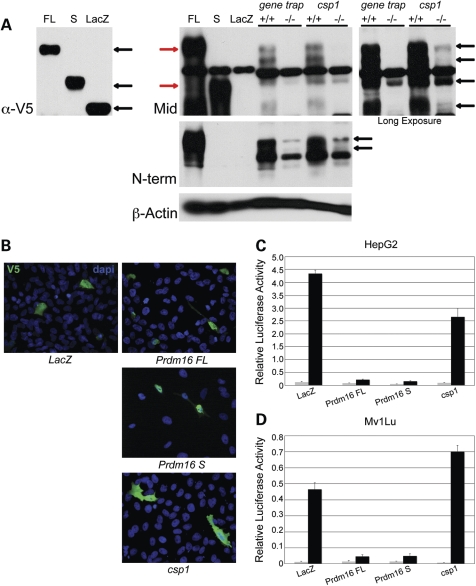
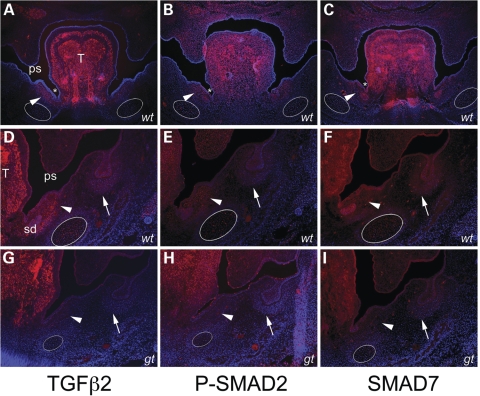
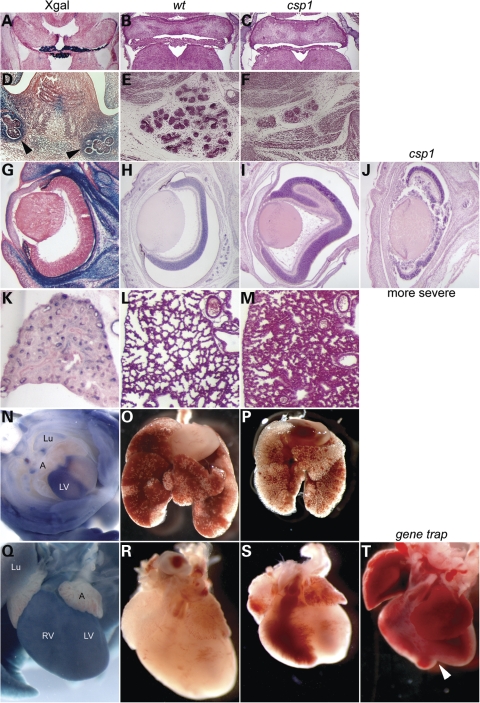
Transcriptional cofactors are essential to the regulation of transforming growth factor beta (TGFbeta) superfamily signaling and play critical and widespread roles during embryonic development, including craniofacial development. We describe the cleft secondary palate 1 (csp1) N-ethyl-N-nitrosourea-induced mouse model of non-syndromic cleft palate (NSCP) that is caused by an intronic Prdm16 splicing mutation. Prdm16 encodes a transcriptional cofactor that regulates TGFbeta signaling, and its expression pattern is consistent with a role in palate and craniofacial development. The cleft palate (CP) appears to be the result of micrognathia and failed palate shelf elevation due to physical obstruction by the tongue, resembling human Pierre Robin sequence (PRS)-like cleft secondary palate. PRDM16 should be considered a candidate for mutation in human clefting disorders, especially NSCP and PRS-like CP.
Figures
References
-
- Mossey P.A., Little J. Epidemiology of oral clefts: an international perspective. In: Wyszynski D.F., editor. Cleft Lip and Palate. From Origin to Treatment. New York: Oxford University Press; 2002. pp. 127–158.
-
- Wyszynski D.F. Cleft Lip and Palate: From Origin to Treatment. 1st edn. Oxford: Oxford University Press; 2002.
-
- FitzPatrick D.R., Carr I.M., McLaren L., Leek J.P., Wightman P., Williamson K., Gautier P., McGill N., Hayward C., Firth H., et al. Identification of SATB2 as the cleft palate gene on 2q32–q33. Hum. Mol. Genet. 2003;12:2491–2501. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
