

Acute T-cell leukemias remain dependent on Notch signaling despite PTEN and INK4A/ARF loss
- PMID: 20008304
- PMCID: PMC2826229
- DOI: 10.1182/blood-2009-04-214718
Acute T-cell leukemias remain dependent on Notch signaling despite PTEN and INK4A/ARF loss
Abstract
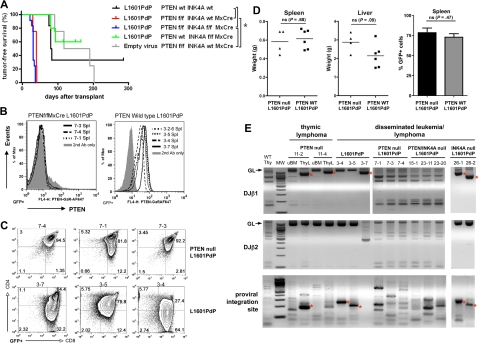
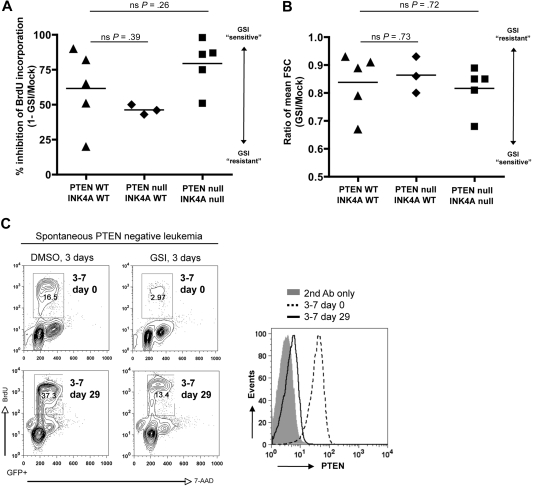
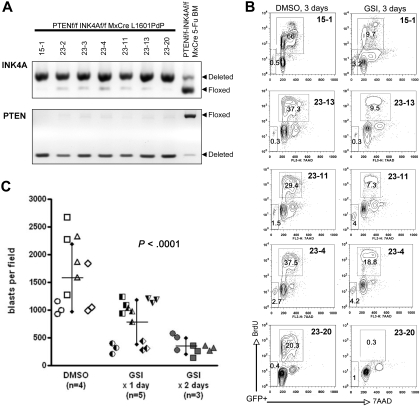
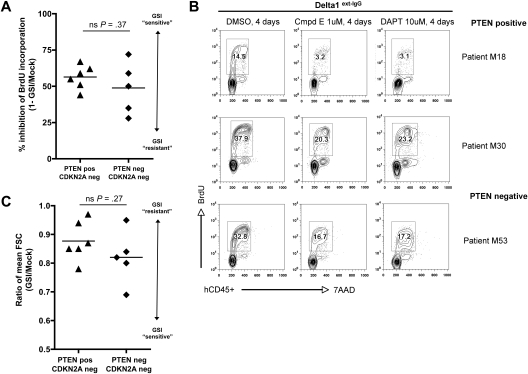
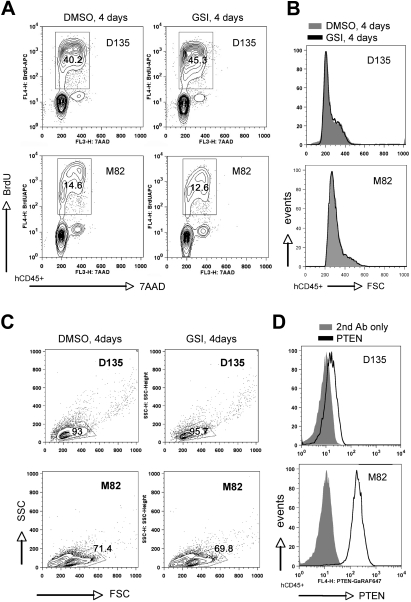
NOTCH1 is activated by mutation in more than 50% of human T-cell acute lymphoblastic leukemias (T-ALLs) and inhibition of Notch signaling causes cell-cycle/growth arrest, providing rationale for NOTCH1 as a therapeutic target. The tumor suppressor phosphatase and tensin homolog (PTEN) is also mutated or lost in up to 20% of cases. It was recently observed among human T-ALL cell lines that PTEN loss correlated with resistance to Notch inhibition, raising concern that patients with PTEN-negative disease may fail Notch inhibitor therapy. As these studies were limited to established cell lines, we addressed this issue using a genetically defined mouse retroviral transduction/bone marrow transplantation model and observed primary murine leukemias to remain dependent on NOTCH1 signaling despite Pten loss, with or without additional deletion of p16(Ink4a)/p19(Arf). We also examined 13 primary human T-ALL samples obtained at diagnosis and found no correlation between PTEN status and resistance to Notch inhibition. Furthermore, we noted in the mouse model that Pten loss accelerated disease onset and produced multiclonal tumors, suggesting NOTCH1 activation and Pten loss may collaborate in leukemia induction. Thus, in contrast to previous findings with established cell lines, these results indicate PTEN loss does not relieve primary T-ALL cells of their "addiction" to Notch signaling.
Figures
References
-
- Maillard I, Fang T, Pear WS. Regulation of lymphoid development, differentiation, and function by the Notch pathway. Annu Rev Immunol. 2005;23:945–974. - PubMed
-
- Grabher C, von Boehmer H, Look AT. Notch 1 activation in the molecular pathogenesis of T-cell acute lymphoblastic leukaemia. Nat Rev Cancer. 2006;6(5):347–359. - PubMed
-
- Weng AP, Ferrando AA, Lee W, et al. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science. 2004;306(5694):269–271. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
