

CK2 phosphorylation-dependent interaction between aprataxin and MDC1 in the DNA damage response
- PMID: 20008512
- PMCID: PMC2836575
- DOI: 10.1093/nar/gkp1149
CK2 phosphorylation-dependent interaction between aprataxin and MDC1 in the DNA damage response
Abstract
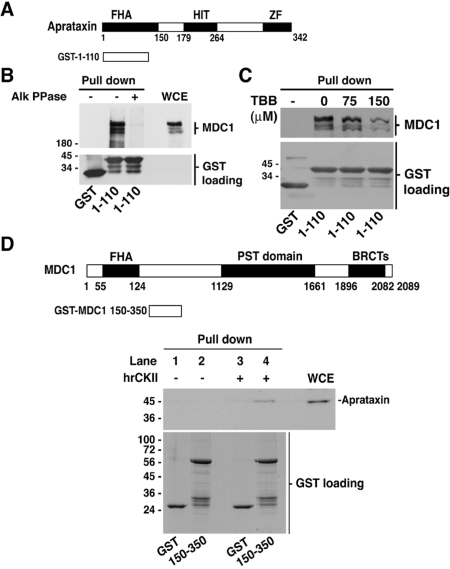
Aprataxin, defective in the neurodegenerative disorder ataxia oculomotor apraxia type 1, resolves abortive DNA ligation intermediates during DNA repair. Here, we demonstrate that aprataxin localizes at sites of DNA damage induced by high LET radiation and binds to mediator of DNA-damage checkpoint protein 1 (MDC1/NFBD1) through a phosphorylation-dependent interaction. This interaction is mediated via the aprataxin FHA domain and multiple casein kinase 2 di-phosphorylated S-D-T-D motifs in MDC1. X-ray structural and mutagenic analysis of aprataxin FHA domain, combined with modelling of the pSDpTD peptide interaction suggest an unusual FHA binding mechanism mediated by a cluster of basic residues at and around the canonical pT-docking site. Mutation of aprataxin FHA Arg29 prevented its interaction with MDC1 and recruitment to sites of DNA damage. These results indicate that aprataxin is involved not only in single strand break repair but also in the processing of a subset of double strand breaks presumably through its interaction with MDC1.
Figures







Similar articles
-
Phospho-dependent interactions between NBS1 and MDC1 mediate chromatin retention of the MRN complex at sites of DNA damage.EMBO Rep. 2008 Aug;9(8):795-801. doi: 10.1038/embor.2008.103. Epub 2008 Jun 27. EMBO Rep. 2008. PMID: 18583988 Free PMC article.
-
MDC1 is a mediator of the mammalian DNA damage checkpoint.Nature. 2003 Feb 27;421(6926):961-6. doi: 10.1038/nature01446. Nature. 2003. PMID: 12607005
-
The molecular basis of ATM-dependent dimerization of the Mdc1 DNA damage checkpoint mediator.Nucleic Acids Res. 2012 May;40(9):3913-28. doi: 10.1093/nar/gkr1300. Epub 2012 Jan 10. Nucleic Acids Res. 2012. PMID: 22234878 Free PMC article.
-
MDC1/NFBD1: a key regulator of the DNA damage response in higher eukaryotes.DNA Repair (Amst). 2004 Aug-Sep;3(8-9):953-7. doi: 10.1016/j.dnarep.2004.03.007. DNA Repair (Amst). 2004. PMID: 15279781 Review.
-
Molecular underpinnings of Aprataxin RNA/DNA deadenylase function and dysfunction in neurological disease.Prog Biophys Mol Biol. 2015 Mar;117(2-3):157-165. doi: 10.1016/j.pbiomolbio.2015.01.007. Epub 2015 Jan 29. Prog Biophys Mol Biol. 2015. PMID: 25637650 Free PMC article. Review.
Cited by
-
Pathway Analysis of Fucoidan Activity Using a Yeast Gene Deletion Library Screen.Mar Drugs. 2019 Jan 14;17(1):54. doi: 10.3390/md17010054. Mar Drugs. 2019. PMID: 30646537 Free PMC article.
-
Discovery of Novel Dual-Target Inhibitor of Bromodomain-Containing Protein 4/Casein Kinase 2 Inducing Apoptosis and Autophagy-Associated Cell Death for Triple-Negative Breast Cancer Therapy.J Med Chem. 2021 Dec 23;64(24):18025-18053. doi: 10.1021/acs.jmedchem.1c01382. Epub 2021 Dec 15. J Med Chem. 2021. PMID: 34908415 Free PMC article.
-
DNA damage response signaling pathways and targets for radiotherapy sensitization in cancer.Signal Transduct Target Ther. 2020 May 1;5(1):60. doi: 10.1038/s41392-020-0150-x. Signal Transduct Target Ther. 2020. PMID: 32355263 Free PMC article. Review.
-
Predicting CK2 beta-dependent substrates using linear patterns.Biochem Biophys Rep. 2015 Aug 20;4:20-27. doi: 10.1016/j.bbrep.2015.08.011. eCollection 2015 Dec. Biochem Biophys Rep. 2015. PMID: 29124183 Free PMC article.
-
Species variations in XRCC1 recruitment strategies for FHA domain-containing proteins.DNA Repair (Amst). 2022 Feb;110:103263. doi: 10.1016/j.dnarep.2021.103263. Epub 2021 Dec 24. DNA Repair (Amst). 2022. PMID: 35026705 Free PMC article.
References
-
- Moreira MC, Barbot C, Tachi N, Kozuka N, Uchida E, Gibson T, Mendonca P, Costa M, Barros J, Yanagisawa T, et al. The gene mutated in ataxia-ocular apraxia type 1 encodes the new HIT/Zn-finger protein aprataxin. Nat. Genet. 2001;29:189–193. - PubMed
-
- Date H, Onodera O, Tanaka H, Iwabuchi K, Uekawa K, Igarashi S, Koike R, Hiroi T, Yuasa T, Awaya Y, et al. Early-onset ataxia with ocular motor apraxia and hypoalbuminemia is caused by mutations in a new HIT superfamily gene. Nat. Genet. 2001;29:184–188. - PubMed
-
- Aicardi J, Barbosa C, Andermann E, Andermann F, Morcos R, Ghanem Q, Fukuyama Y, Awaya Y, Moe P. Ataxia-ocular motor apraxia: asyndrome mimicking ataxia-telangiectasia. Ann. Neurol. 1988;4:497–502. - PubMed
-
- Lavin MF, Shiloh Y. The genetic defect in ataxia-telangiectasia. Annu. Rev. Immunol. 1997;15:177–202. - PubMed
-
- Lavin MF, Kozlov S. ATM activation and DNA damage response. Cell Cycle. 2007;6:931–942. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
