

Klebsiella pneumoniae capsule polysaccharide impedes the expression of beta-defensins by airway epithelial cells
- PMID: 20008534
- PMCID: PMC2825953
- DOI: 10.1128/IAI.00940-09
Klebsiella pneumoniae capsule polysaccharide impedes the expression of beta-defensins by airway epithelial cells
Erratum in
- Infect Immun. 2010 Dec;78(12):5352. Larrate, Eider [corrected to Larrarte, Eider]
Abstract
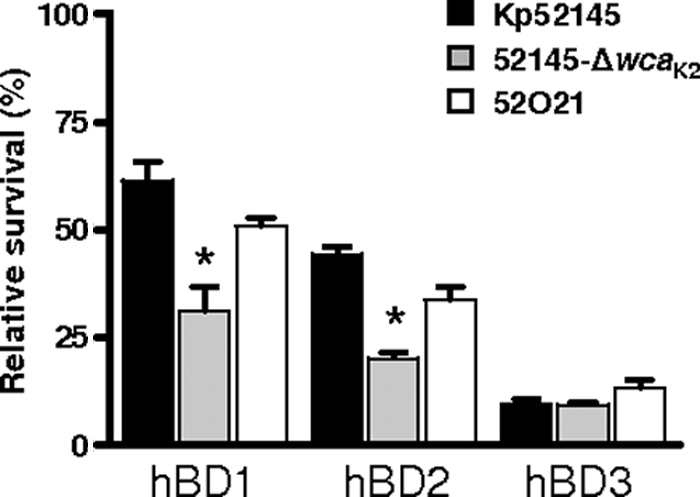
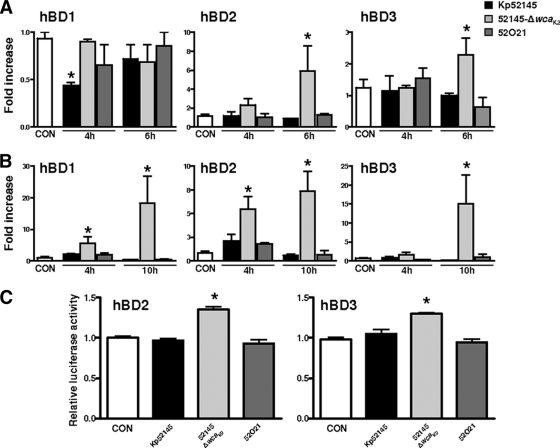
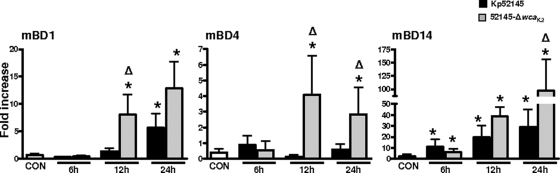
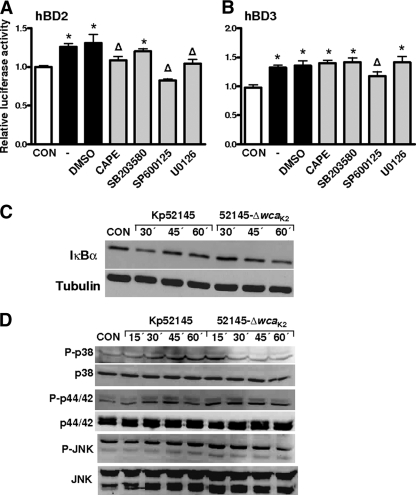
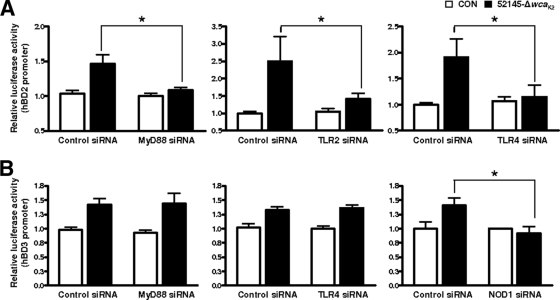
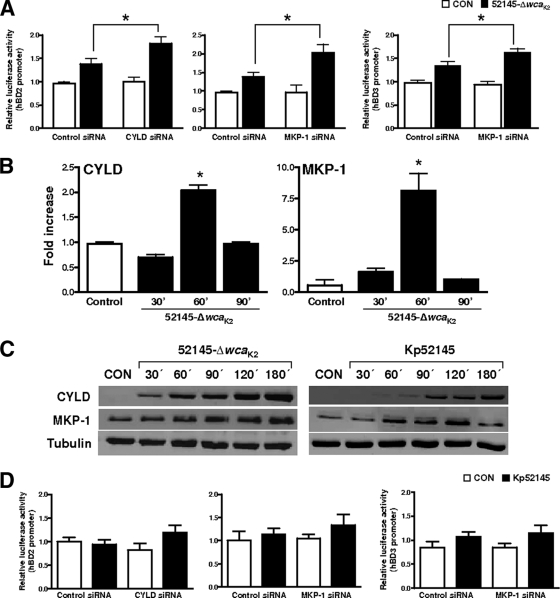
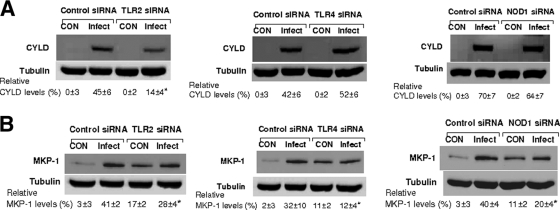
Human beta-defensins (hBDs) contribute to the protection of the respiratory tract against pathogens. It is reasonable to postulate that pathogens have developed countermeasures to resist them. Klebsiella pneumoniae capsule polysaccharide (CPS), but not the lipopolysaccharide O antigen, mediated resistance against hBD1 and hBD2. hBD3 was the most potent hBD against Klebsiella. We investigated the possibility that as a strategy for survival in the lung, K. pneumoniae may not activate the expression of hBDs. Infection of A549 and normal human bronchial cells with 52145-Deltawca(K2), a CPS mutant, increased the expression of hBD2 and hBD3. Neither the wild type nor the lipopolysaccharide O antigen mutant increased the expression of hBDs. In vivo, 52145-Deltawca(K2) induced higher levels of mBD4 and mBD14, possible mouse orthologues of hBD2 and hBD3, respectively, than the wild type. 52145-Deltawca(K2)-dependent upregulation of hBD2 occurred via NF-kappaB and mitogen-activated protein kinases (MAPKs) p44/42, Jun N-terminal protein kinase (JNK)-dependent pathways. The increase in hBD3 expression was dependent on the MAPK JNK. 52145-Deltawca(K2) engaged Toll-like receptors 2 and 4 (TLR2 and TLR4) to activate hBD2, whereas hBD3 expression was dependent on NOD1. K. pneumoniae induced the expression of CYLD and MKP-1, which act as negative regulators for 52145-Deltawca(K2)-induced expression of hBDs. Bacterial engagement of pattern recognition receptors induced CYLD and MKP-1, which may initiate the attenuation of proinflammatory pathways. The results of this study indicate that K. pneumoniae CPS not only protects the pathogen from the bactericidal action of defensins but also impedes their expression. These features of K. pneumoniae CPS may facilitate pathogen survival in the hostile environment of the lung.
Figures
Similar articles
-
Klebsiella pneumoniae outer membrane protein A is required to prevent the activation of airway epithelial cells.J Biol Chem. 2011 Mar 25;286(12):9956-67. doi: 10.1074/jbc.M110.181008. Epub 2011 Jan 28. J Biol Chem. 2011. PMID: 21278256 Free PMC article.
-
Klebsiella pneumoniae increases the levels of Toll-like receptors 2 and 4 in human airway epithelial cells.Infect Immun. 2009 Feb;77(2):714-24. doi: 10.1128/IAI.00852-08. Epub 2008 Nov 17. Infect Immun. 2009. PMID: 19015258 Free PMC article.
-
Klebsiella pneumoniae subverts the activation of inflammatory responses in a NOD1-dependent manner.Cell Microbiol. 2011 Jan;13(1):135-53. doi: 10.1111/j.1462-5822.2010.01526.x. Epub 2010 Oct 14. Cell Microbiol. 2011. PMID: 20846183
-
Nucleotide-binding oligomerization domain-1 and epidermal growth factor receptor: critical regulators of beta-defensins during Helicobacter pylori infection.J Biol Chem. 2006 Apr 28;281(17):11637-48. doi: 10.1074/jbc.M510275200. Epub 2006 Mar 2. J Biol Chem. 2006. PMID: 16513653
-
Klebsiella pneumoniae targets an EGF receptor-dependent pathway to subvert inflammation.Cell Microbiol. 2013 Jul;15(7):1212-33. doi: 10.1111/cmi.12110. Epub 2013 Feb 17. Cell Microbiol. 2013. PMID: 23347154
Cited by
-
A trans-kingdom T6SS effector induces the fragmentation of the mitochondrial network and activates innate immune receptor NLRX1 to promote infection.Nat Commun. 2023 Feb 16;14(1):871. doi: 10.1038/s41467-023-36629-3. Nat Commun. 2023. PMID: 36797302 Free PMC article.
-
Klebsiella pneumoniae ST258 Negatively Regulates the Oxidative Burst in Human Neutrophils.Front Immunol. 2019 Apr 26;10:929. doi: 10.3389/fimmu.2019.00929. eCollection 2019. Front Immunol. 2019. PMID: 31105712 Free PMC article.
-
Innate Immune Signaling Activated by MDR Bacteria in the Airway.Physiol Rev. 2016 Jan;96(1):19-53. doi: 10.1152/physrev.00009.2015. Physiol Rev. 2016. PMID: 26582515 Free PMC article. Review.
-
Elucidation of the RamA regulon in Klebsiella pneumoniae reveals a role in LPS regulation.PLoS Pathog. 2015 Jan 29;11(1):e1004627. doi: 10.1371/journal.ppat.1004627. eCollection 2015 Jan. PLoS Pathog. 2015. PMID: 25633080 Free PMC article.
-
Dissection of host cell signal transduction during Acinetobacter baumannii-triggered inflammatory response.PLoS One. 2010 Apr 7;5(4):e10033. doi: 10.1371/journal.pone.0010033. PLoS One. 2010. PMID: 20383325 Free PMC article.
References
-
- Aarbiou, J., K. F. Rabe, and P. S. Hiemstra. 2002. Role of defensins in inflammatory lung disease. Ann. Med. 34:96-101. - PubMed
-
- Aderem, A., and R. J. Ulevitch. 2000. Toll-like receptors in the induction of the innate immune response. Nature 406:782-787. - PubMed
-
- Agerberth, B., J. Grunewald, E. Castanos-Velez, B. Olsson, H. Jornvall, H. Wigzell, A. Eklund, and G. H. Gudmundsson. 1999. Antibacterial components in bronchoalveolar lavage fluid from healthy individuals and sarcoidosis patients. Am. J. Respir. Crit. Care Med. 160:283-290. - PubMed
-
- Akira, S., S. Uematsu, and O. Takeuchi. 2006. Pathogen recognition and innate immunity. Cell 124:783-801. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
