

Muscle inactivation of mTOR causes metabolic and dystrophin defects leading to severe myopathy
- PMID: 20008564
- PMCID: PMC2806319
- DOI: 10.1083/jcb.200903131
Muscle inactivation of mTOR causes metabolic and dystrophin defects leading to severe myopathy
Abstract
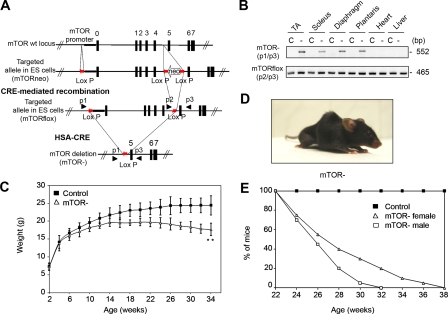
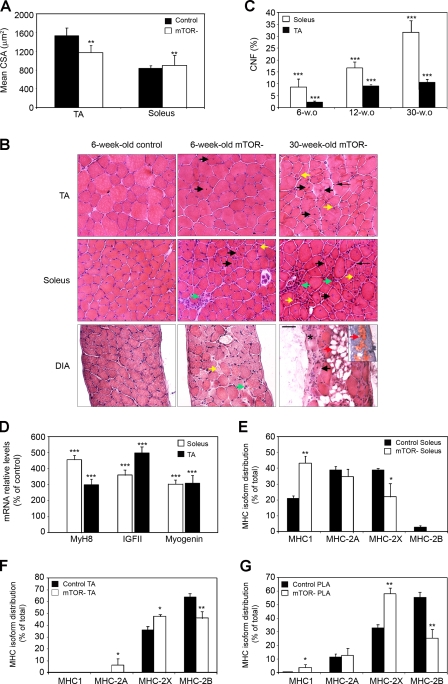
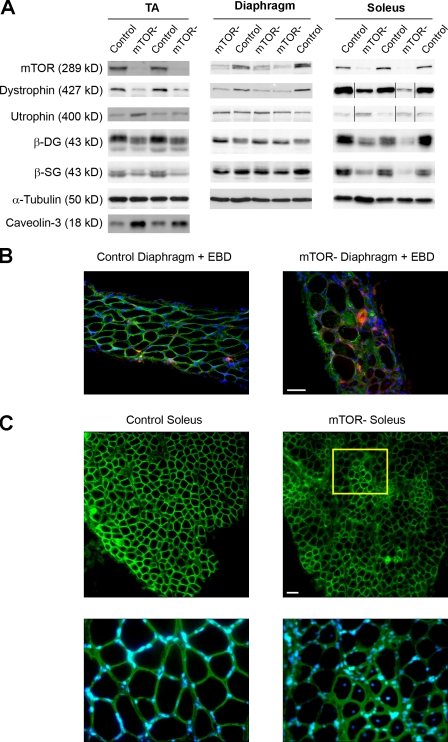
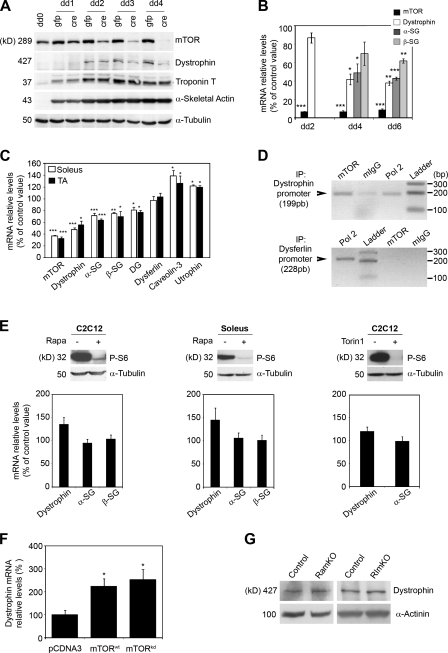
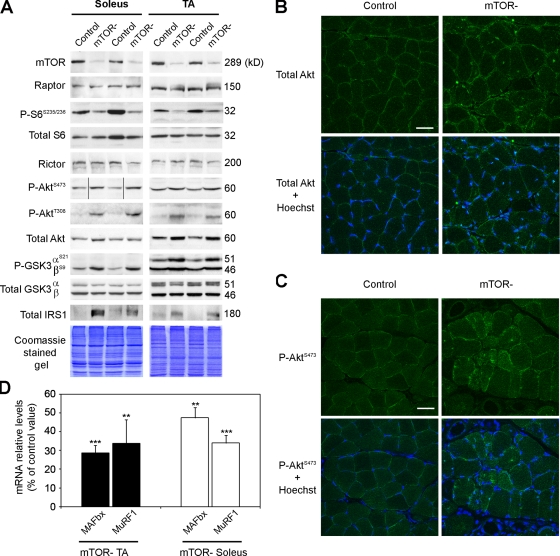
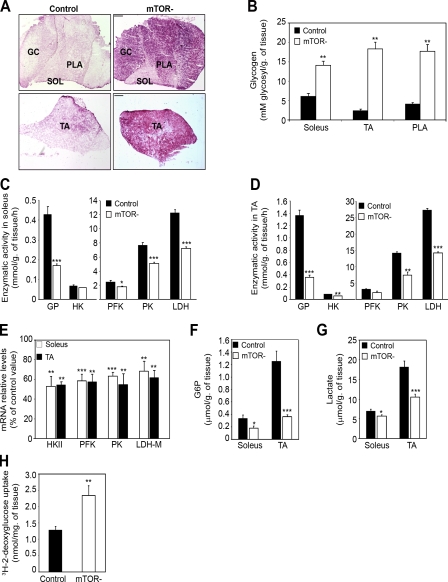
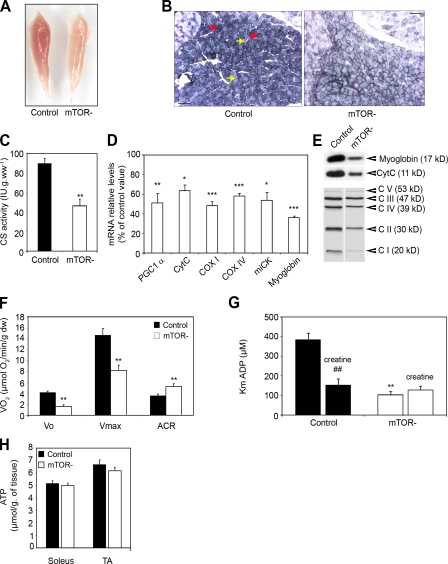
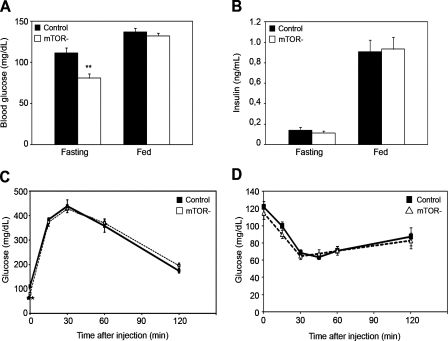
Mammalian target of rapamycin (mTOR) is a key regulator of cell growth that associates with raptor and rictor to form the mTOR complex 1 (mTORC1) and mTORC2, respectively. Raptor is required for oxidative muscle integrity, whereas rictor is dispensable. In this study, we show that muscle-specific inactivation of mTOR leads to severe myopathy, resulting in premature death. mTOR-deficient muscles display metabolic changes similar to those observed in muscles lacking raptor, including impaired oxidative metabolism, altered mitochondrial regulation, and glycogen accumulation associated with protein kinase B/Akt hyperactivation. In addition, mTOR-deficient muscles exhibit increased basal glucose uptake, whereas whole body glucose homeostasis is essentially maintained. Importantly, loss of mTOR exacerbates the myopathic features in both slow oxidative and fast glycolytic muscles. Moreover, mTOR but not raptor and rictor deficiency leads to reduced muscle dystrophin content. We provide evidence that mTOR controls dystrophin transcription in a cell-autonomous, rapamycin-resistant, and kinase-independent manner. Collectively, our results demonstrate that mTOR acts mainly via mTORC1, whereas regulation of dystrophin is raptor and rictor independent.
Figures
Similar articles
-
mTORC1 and mTORC2 regulate insulin secretion through Akt in INS-1 cells.J Endocrinol. 2013 Jan 2;216(1):21-9. doi: 10.1530/JOE-12-0351. Print 2013 Jan. J Endocrinol. 2013. PMID: 23092880
-
Skeletal muscle-specific ablation of raptor, but not of rictor, causes metabolic changes and results in muscle dystrophy.Cell Metab. 2008 Nov;8(5):411-24. doi: 10.1016/j.cmet.2008.10.002. Cell Metab. 2008. PMID: 19046572
-
Enhancing mammalian target of rapamycin (mTOR)-targeted cancer therapy by preventing mTOR/raptor inhibition-initiated, mTOR/rictor-independent Akt activation.Cancer Res. 2008 Sep 15;68(18):7409-18. doi: 10.1158/0008-5472.CAN-08-1522. Cancer Res. 2008. PMID: 18794129 Free PMC article.
-
The mTORC2 complex regulates terminal differentiation of C2C12 myoblasts.Mol Cell Biol. 2009 Sep;29(17):4691-700. doi: 10.1128/MCB.00764-09. Epub 2009 Jun 29. Mol Cell Biol. 2009. PMID: 19564418 Free PMC article.
-
mTOR couples cellular nutrient sensing to organismal metabolic homeostasis.Trends Endocrinol Metab. 2011 Mar;22(3):94-102. doi: 10.1016/j.tem.2010.12.003. Epub 2011 Jan 25. Trends Endocrinol Metab. 2011. PMID: 21269838 Free PMC article. Review.
Cited by
-
Defective mitochondrial morphology and bioenergetic function in mice lacking the transcription factor Yin Yang 1 in skeletal muscle.Mol Cell Biol. 2012 Aug;32(16):3333-46. doi: 10.1128/MCB.00337-12. Epub 2012 Jun 18. Mol Cell Biol. 2012. PMID: 22711985 Free PMC article.
-
Signaling Pathways That Control Muscle Mass.Int J Mol Sci. 2020 Jul 4;21(13):4759. doi: 10.3390/ijms21134759. Int J Mol Sci. 2020. PMID: 32635462 Free PMC article. Review.
-
mTOR Regulates Gap Junction Alpha-1 Protein Trafficking in Sertoli Cells and Is Required for the Maintenance of Spermatogenesis in Mice.Biol Reprod. 2016 Jul;95(1):13. doi: 10.1095/biolreprod.115.138016. Epub 2016 Jun 8. Biol Reprod. 2016. PMID: 27281705 Free PMC article.
-
Ginsenoside Rg5 promotes muscle regeneration via p38MAPK and Akt/mTOR signaling.J Ginseng Res. 2023 Nov;47(6):726-734. doi: 10.1016/j.jgr.2023.06.004. Epub 2023 Jun 24. J Ginseng Res. 2023. PMID: 38107401 Free PMC article.
-
Rapamycin Exacerbates Staphylococcus aureus Pneumonia by Inhibiting mTOR-RPS6 in Macrophages.J Inflamm Res. 2023 Nov 30;16:5715-5728. doi: 10.2147/JIR.S434483. eCollection 2023. J Inflamm Res. 2023. PMID: 38053607 Free PMC article.
References
-
- Acharyya S., Butchbach M.E., Sahenk Z., Wang H., Saji M., Carathers M., Ringel M.D., Skipworth R.J., Fearon K.C., Hollingsworth M.A., et al. 2005. Dystrophin glycoprotein complex dysfunction: a regulatory link between muscular dystrophy and cancer cachexia. Cancer Cell. 8:421–432 10.1016/j.ccr.2005.10.004 - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
