

Alpha1-syntrophin mutations identified in sudden infant death syndrome cause an increase in late cardiac sodium current
- PMID: 20009079
- PMCID: PMC2810855
- DOI: 10.1161/CIRCEP.109.891440
Alpha1-syntrophin mutations identified in sudden infant death syndrome cause an increase in late cardiac sodium current
Abstract
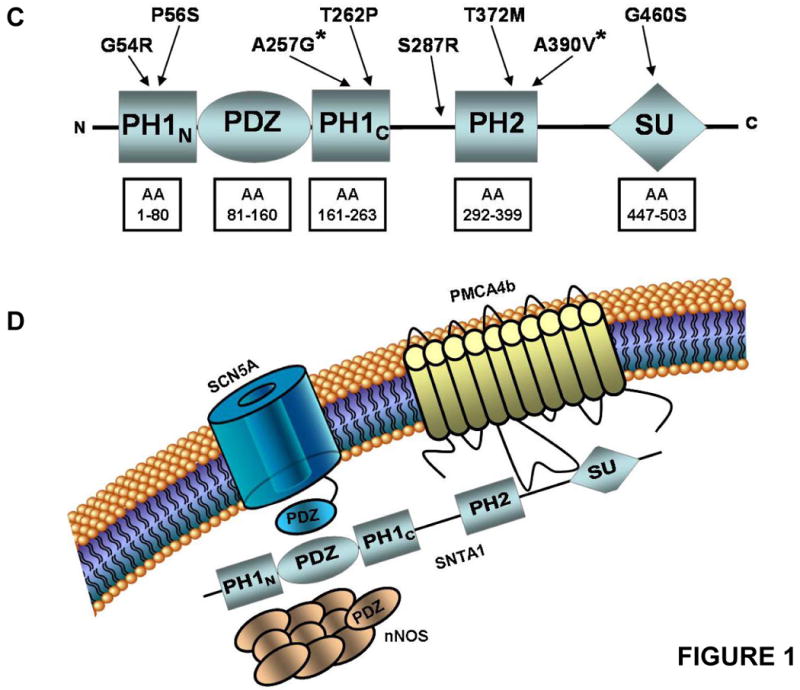
Background: Sudden infant death syndrome (SIDS) is a leading cause of death during the first 6 months after birth. About 5% to 10% of SIDS may stem from cardiac channelopathies such as long-QT syndrome. We recently implicated mutations in alpha1-syntrophin (SNTA1) as a novel cause of long-QT syndrome, whereby mutant SNTA1 released inhibition of associated neuronal nitric oxide synthase by the plasma membrane Ca-ATPase PMCA4b, causing increased peak and late sodium current (I(Na)) via S-nitrosylation of the cardiac sodium channel. This study determined the prevalence and functional properties of SIDS-associated SNTA1 mutations.
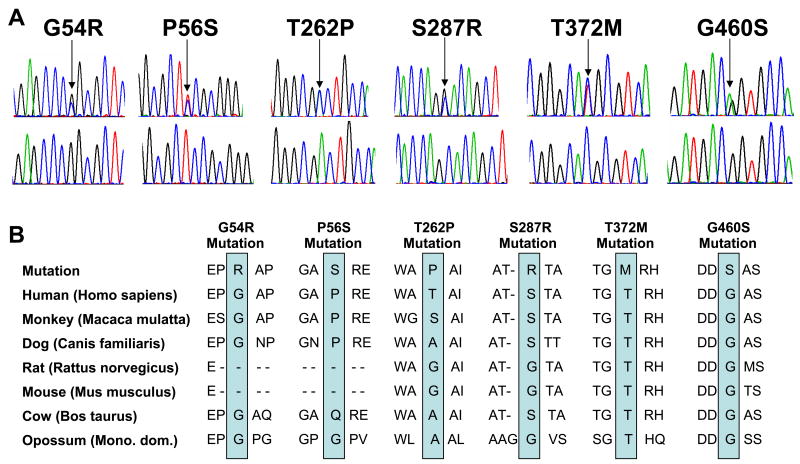
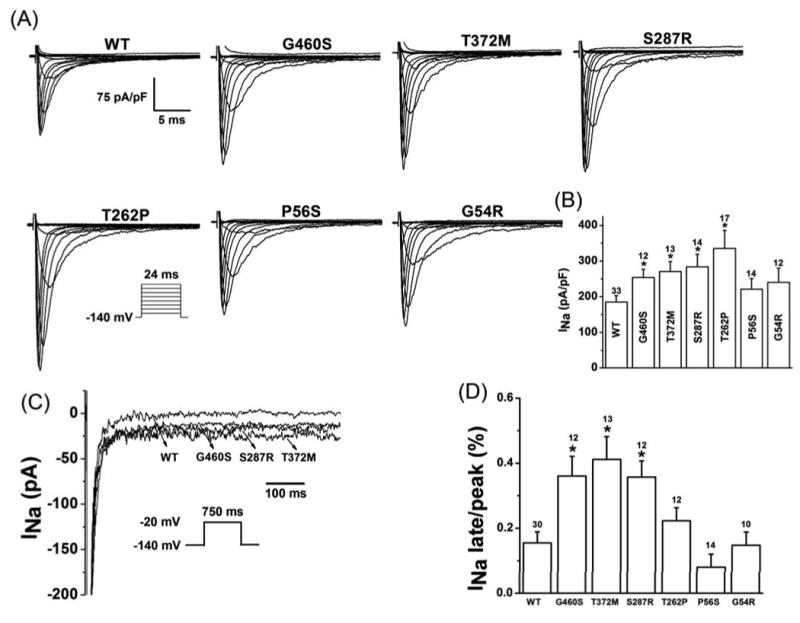
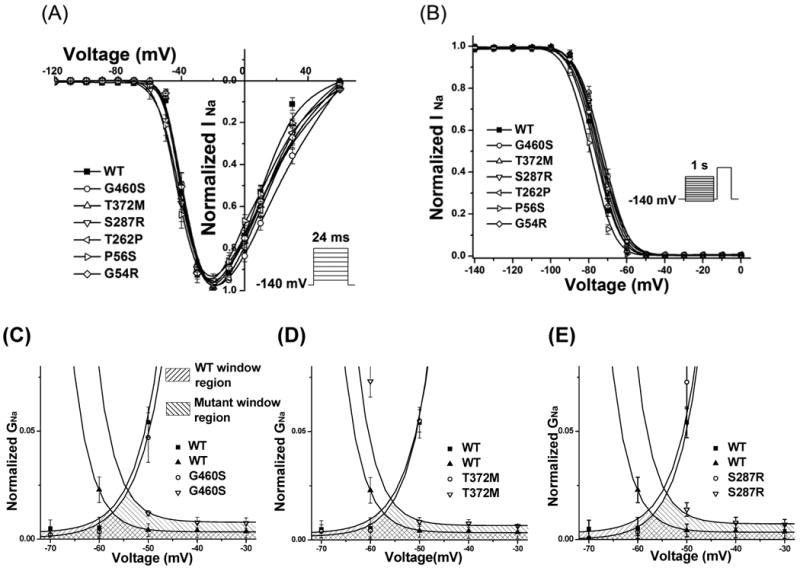
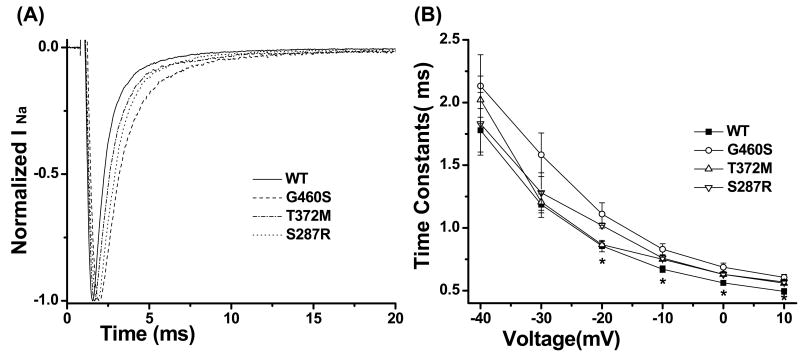
Methods and results: Using polymerase chain reaction, denaturing high-performance liquid chromatography, and DNA sequencing of SNTA1's open reading frame, 6 rare (absent in 800 reference alleles) missense mutations (G54R, P56S, T262P, S287R, T372M, and G460S) were identified in 8 (approximately 3%) of 292 SIDS cases. These mutations were engineered using polymerase chain reaction-based overlap extension and were coexpressed heterologously with SCN5A, neuronal nitric oxide synthase, and PMCA4b in HEK293 cells. I(Na) was recorded using the whole-cell method. A significant 1.4- to 1.5-fold increase in peak I(Na) and 2.3- to 2.7-fold increase in late I(Na) compared with controls was evident for S287R-, T372M-, and G460S-SNTA1 and was reversed by a neuronal nitric oxide synthase inhibitor. These 3 mutations also caused a significant depolarizing shift in channel inactivation, thereby increasing the overlap of the activation and inactivation curves to increase window current.
Conclusions: Abnormal biophysical phenotypes implicate mutations in SNTA1 as a novel pathogenic mechanism for the subset of channelopathic SIDS. Functional studies are essential to distinguish pathogenic perturbations in channel interacting proteins such as alpha1-syntrophin from similarly rare but innocuous ones.
Figures
References
-
- Krous HF, Beckwith JB, Byard RW, Rognum TO, Bajanowski T, Corey T, Cutz E, Hanzlick R, Keens TG, Mitchell EA. Sudden infant death syndrome and unclassified sudden infant deaths: a definitional and diagnostic approach. Pediatrics. 2004;114:234–238. - PubMed
-
- Hannah CK. Brainstem mechanisms underlying the sudden infant death syndrome: evidence from human pathologic studies. Dev Psychobiol. 2009;51:223–233. - PubMed
-
- Malloy MH. SIDS -- A syndrome in search of a cause. N Engl J Med. 2004;351:957–959. - PubMed
-
- Mitchell EA. What is the mechanism of SIDS? Clues from epidemiology. Dev Psychobiol. 2009;51:215–222. - PubMed
-
- Moon RY, Horne RSC, Hauck FR. Sudden infant death syndrome. Lancet. 2007;370:1578–1587. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
