

Label-free, normalized quantification of complex mass spectrometry data for proteomic analysis
- PMID: 20010810
- PMCID: PMC2805705
- DOI: 10.1038/nbt.1592
Label-free, normalized quantification of complex mass spectrometry data for proteomic analysis
Abstract
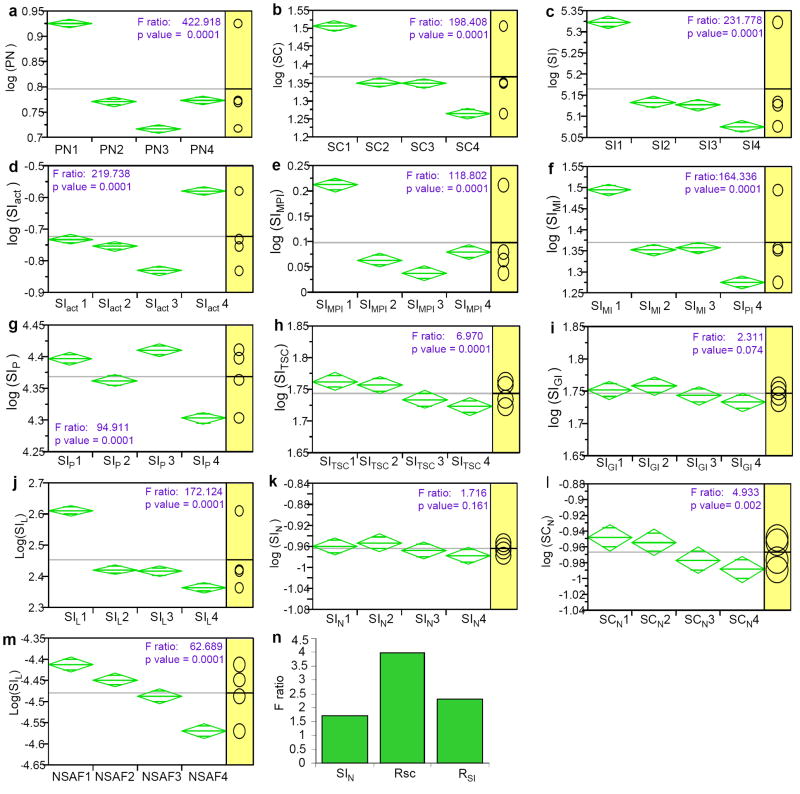
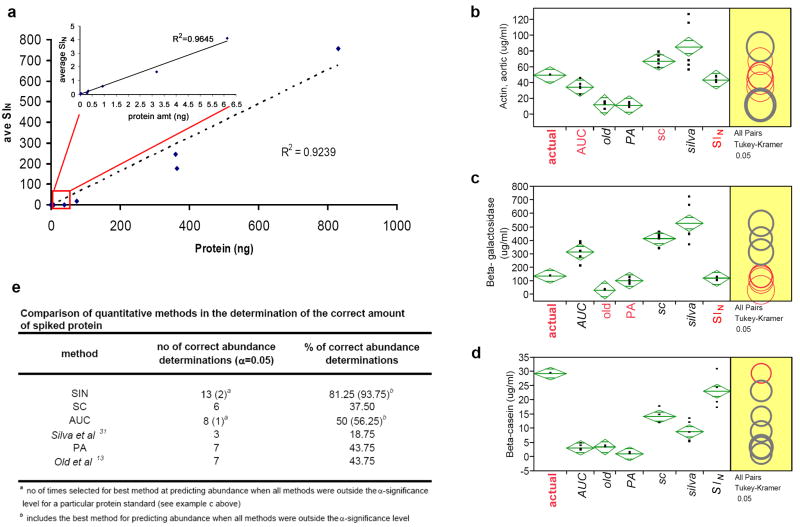
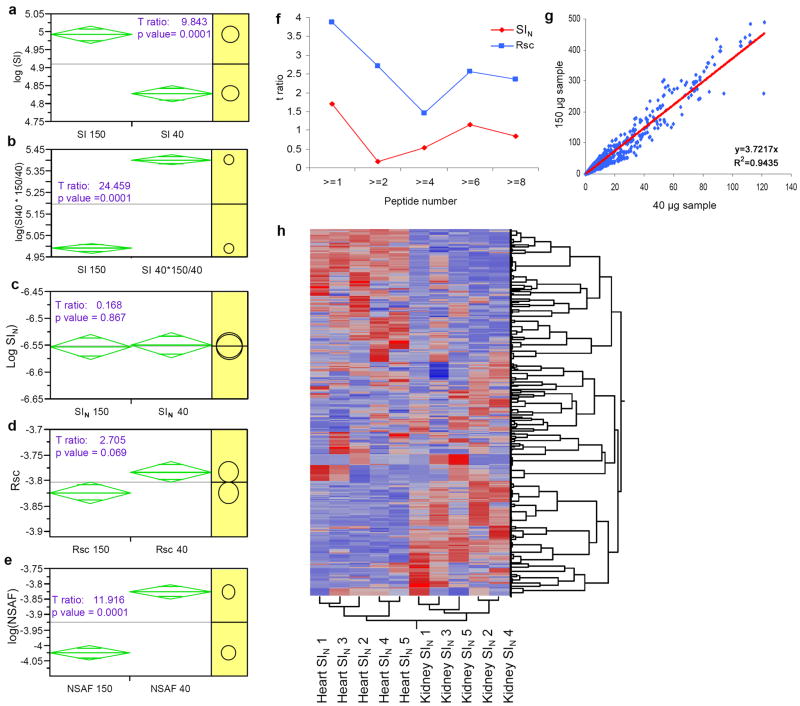
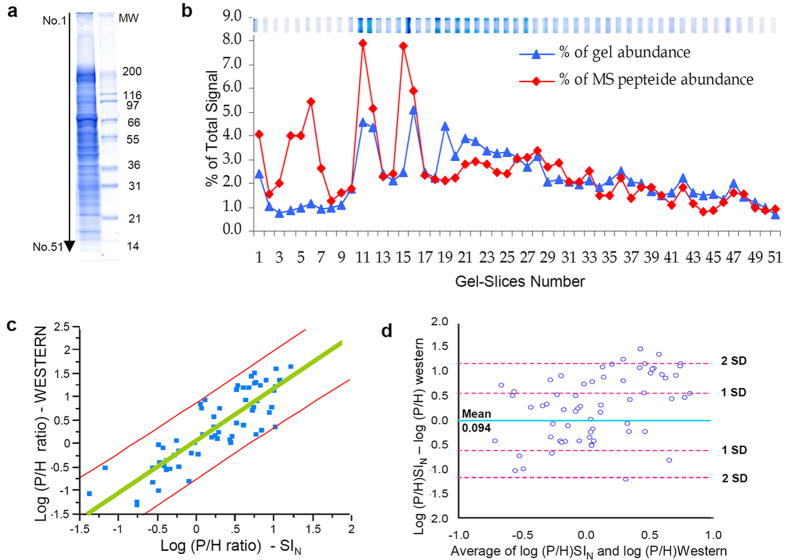
Replicate mass spectrometry (MS) measurements and the use of multiple analytical methods can greatly expand the comprehensiveness of shotgun proteomic profiling of biological samples. However, the inherent biases and variations in such data create computational and statistical challenges for quantitative comparative analysis. We developed and tested a normalized, label-free quantitative method termed the normalized spectral index (SI(N)), which combines three MS abundance features: peptide count, spectral count and fragment-ion (tandem MS or MS/MS) intensity. SI(N) largely eliminated variances between replicate MS measurements, permitting quantitative reproducibility and highly significant quantification of thousands of proteins detected in replicate MS measurements of the same and distinct samples. It accurately predicts protein abundance more often than the five other methods we tested. Comparative immunoblotting and densitometry further validate our method. Comparative quantification of complex data sets from multiple shotgun proteomics measurements is relevant for systems biology and biomarker discovery.
Figures
Similar articles
-
A multi-model statistical approach for proteomic spectral count quantitation.J Proteomics. 2016 Jul 20;144:23-32. doi: 10.1016/j.jprot.2016.05.032. Epub 2016 May 31. J Proteomics. 2016. PMID: 27260494 Free PMC article.
-
Quantification of proteins by label-free LC-MS/MS.Methods Mol Biol. 2010;658:217-31. doi: 10.1007/978-1-60761-780-8_13. Methods Mol Biol. 2010. PMID: 20839107
-
Comparative analysis of statistical methods used for detecting differential expression in label-free mass spectrometry proteomics.J Proteomics. 2015 Nov 3;129:83-92. doi: 10.1016/j.jprot.2015.07.012. Epub 2015 Jul 18. J Proteomics. 2015. PMID: 26193490
-
Mass spectrometry-based label-free quantitative proteomics.J Biomed Biotechnol. 2010;2010:840518. doi: 10.1155/2010/840518. Epub 2009 Nov 10. J Biomed Biotechnol. 2010. PMID: 19911078 Free PMC article. Review.
-
The proteomic advantage: label-free quantification of proteins expressed in bovine milk during experimentally induced coliform mastitis.Vet Immunol Immunopathol. 2010 Dec 15;138(4):252-66. doi: 10.1016/j.vetimm.2010.10.004. Epub 2010 Oct 14. Vet Immunol Immunopathol. 2010. PMID: 21067814 Review.
Cited by
-
Proteomic analysis of the supernatant of red blood cell units: the effects of storage and leucoreduction.Vox Sang. 2013 Oct;105(3):210-8. doi: 10.1111/vox.12042. Epub 2013 May 11. Vox Sang. 2013. PMID: 23663258 Free PMC article.
-
Urinary Apolipoprotein C3 Is a Potential Biomarker for Alzheimer's Disease.Dement Geriatr Cogn Dis Extra. 2020 Sep 11;10(3):94-104. doi: 10.1159/000509561. eCollection 2020 Sep-Dec. Dement Geriatr Cogn Dis Extra. 2020. PMID: 33082773 Free PMC article.
-
Metaproteomics-informed stoichiometric modeling reveals the responses of wetland microbial communities to oxygen and sulfate exposure.NPJ Biofilms Microbiomes. 2024 Jul 3;10(1):55. doi: 10.1038/s41522-024-00525-5. NPJ Biofilms Microbiomes. 2024. PMID: 38961111 Free PMC article.
-
Peptimetric: Quantifying and Visualizing Differences in Peptidomic Data.Front Bioinform. 2021 Aug 25;1:722466. doi: 10.3389/fbinf.2021.722466. eCollection 2021. Front Bioinform. 2021. PMID: 36303760 Free PMC article.
-
Stable association of RNAi machinery is conserved between the cytoplasm and nucleus of human cells.RNA. 2016 Jul;22(7):1085-98. doi: 10.1261/rna.056499.116. Epub 2016 May 19. RNA. 2016. PMID: 27198507 Free PMC article.
References
-
- Durr E, et al. Direct proteomic mapping of the lung microvascular endothelial cell surface in vivo and in cell culture. Nat Biotechnol. 2004;22:985–992. - PubMed
-
- Oh P, et al. Subtractive proteomic mapping of the endothelial surface in lung and solid tumours for tissue-specific therapy. Nature. 2004;429:629–635. - PubMed
-
- Wong JW, Sullivan MJ, Cagney G. Computational methods for the comparative quantification of proteins in label-free LCn-MS experiments. Brief Bioinform. 2008;9:156–165. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
