

The GAAS metagenomic tool and its estimations of viral and microbial average genome size in four major biomes
- PMID: 20011103
- PMCID: PMC2781106
- DOI: 10.1371/journal.pcbi.1000593
The GAAS metagenomic tool and its estimations of viral and microbial average genome size in four major biomes
Abstract
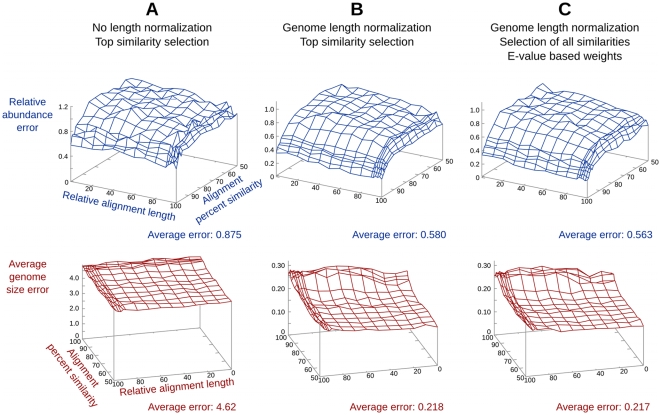
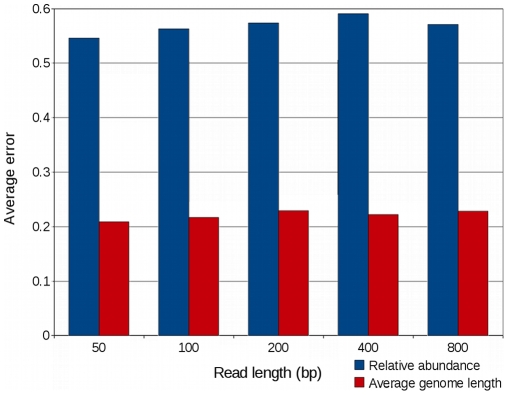
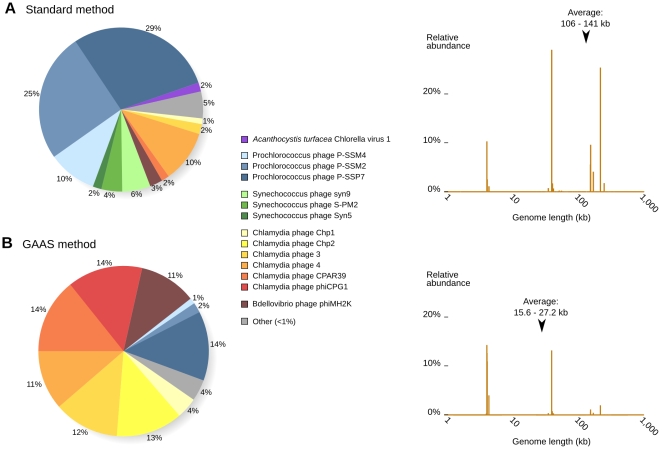
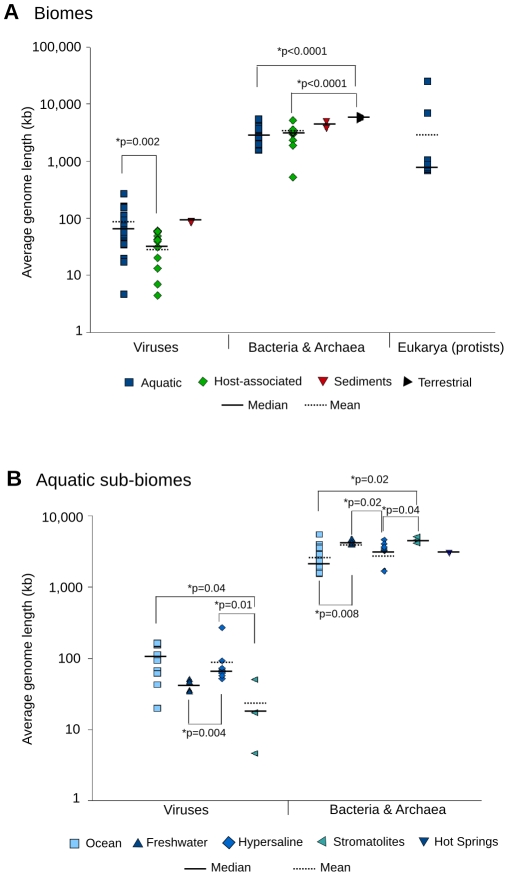
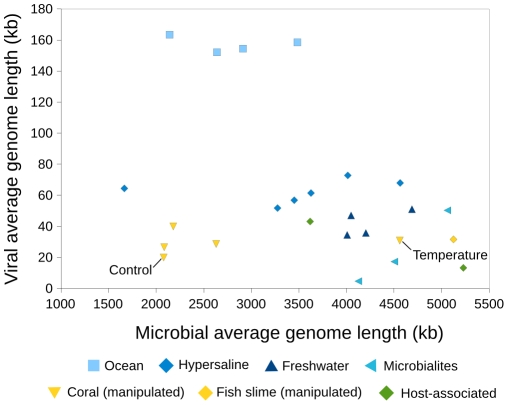
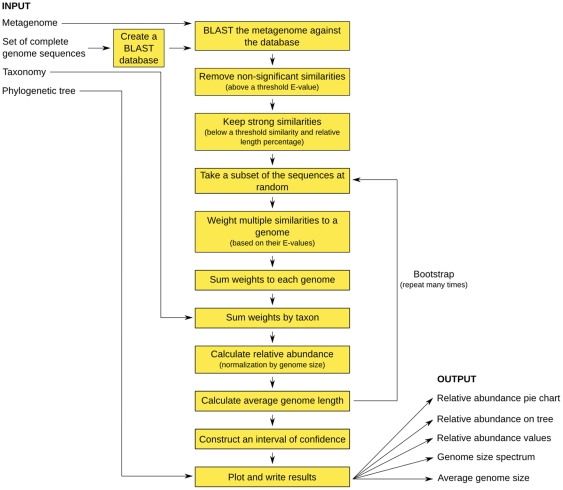
Metagenomic studies characterize both the composition and diversity of uncultured viral and microbial communities. BLAST-based comparisons have typically been used for such analyses; however, sampling biases, high percentages of unknown sequences, and the use of arbitrary thresholds to find significant similarities can decrease the accuracy and validity of estimates. Here, we present Genome relative Abundance and Average Size (GAAS), a complete software package that provides improved estimates of community composition and average genome length for metagenomes in both textual and graphical formats. GAAS implements a novel methodology to control for sampling bias via length normalization, to adjust for multiple BLAST similarities by similarity weighting, and to select significant similarities using relative alignment lengths. In benchmark tests, the GAAS method was robust to both high percentages of unknown sequences and to variations in metagenomic sequence read lengths. Re-analysis of the Sargasso Sea virome using GAAS indicated that standard methodologies for metagenomic analysis may dramatically underestimate the abundance and importance of organisms with small genomes in environmental systems. Using GAAS, we conducted a meta-analysis of microbial and viral average genome lengths in over 150 metagenomes from four biomes to determine whether genome lengths vary consistently between and within biomes, and between microbial and viral communities from the same environment. Significant differences between biomes and within aquatic sub-biomes (oceans, hypersaline systems, freshwater, and microbialites) suggested that average genome length is a fundamental property of environments driven by factors at the sub-biome level. The behavior of paired viral and microbial metagenomes from the same environment indicated that microbial and viral average genome sizes are independent of each other, but indicative of community responses to stressors and environmental conditions.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
