

An abundance of ubiquitously expressed genes revealed by tissue transcriptome sequence data
- PMID: 20011106
- PMCID: PMC2781110
- DOI: 10.1371/journal.pcbi.1000598
An abundance of ubiquitously expressed genes revealed by tissue transcriptome sequence data
Abstract
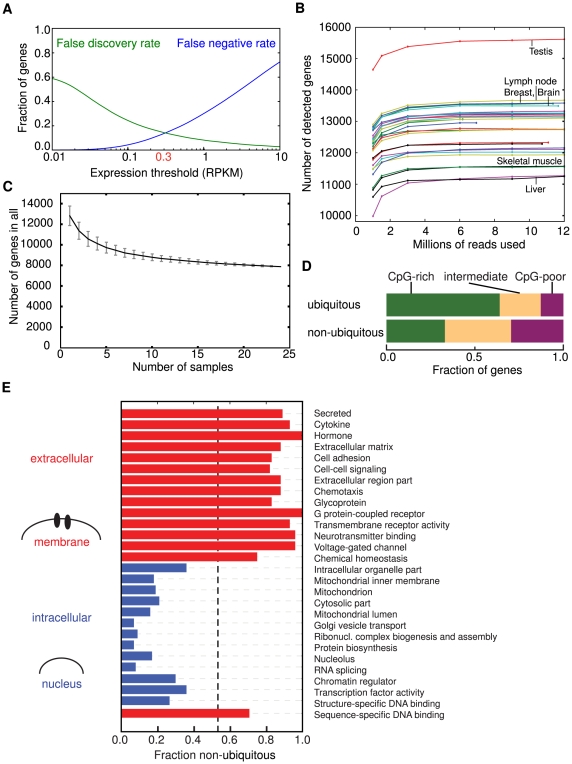
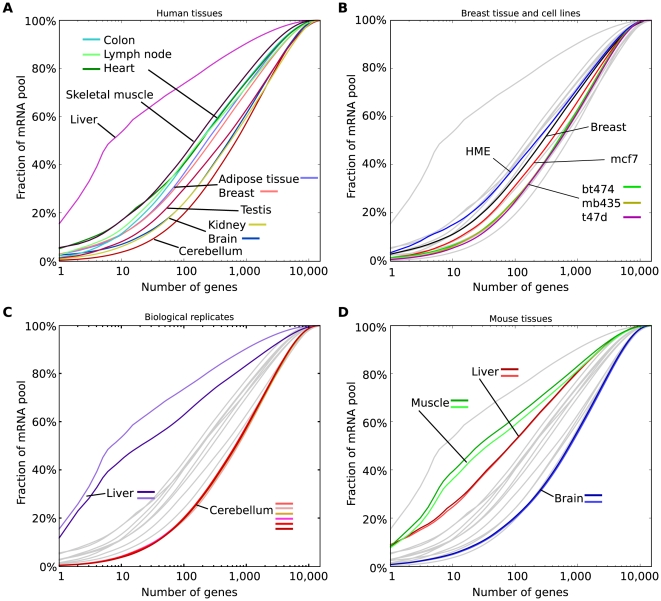
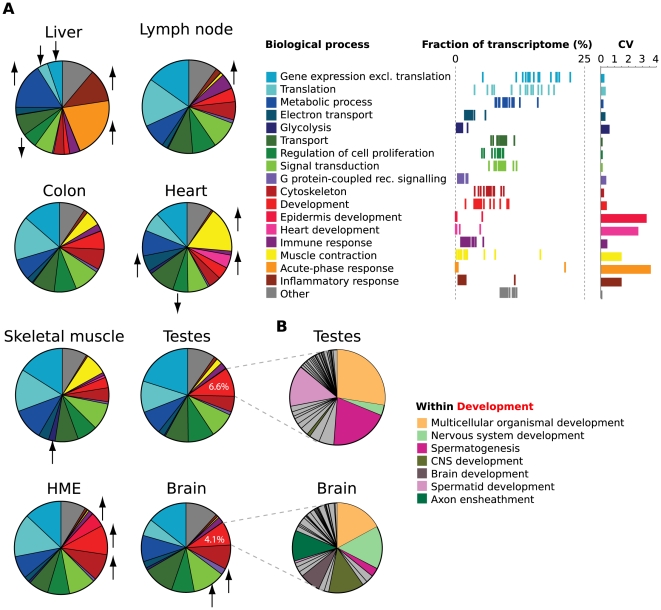
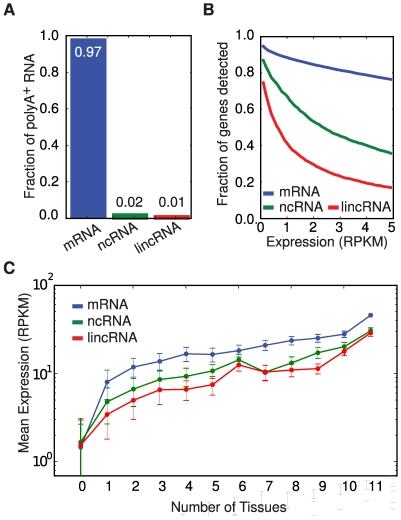
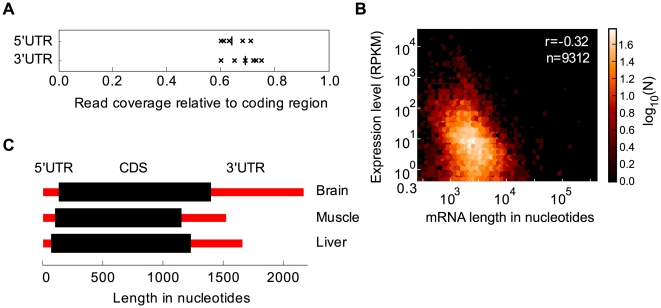
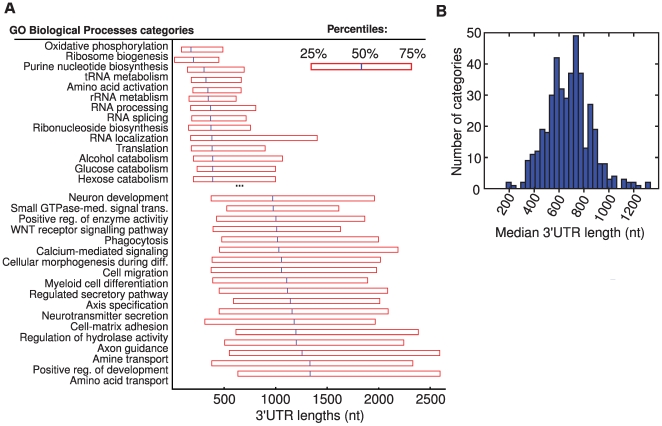
The parts of the genome transcribed by a cell or tissue reflect the biological processes and functions it carries out. We characterized the features of mammalian tissue transcriptomes at the gene level through analysis of RNA deep sequencing (RNA-Seq) data across human and mouse tissues and cell lines. We observed that roughly 8,000 protein-coding genes were ubiquitously expressed, contributing to around 75% of all mRNAs by message copy number in most tissues. These mRNAs encoded proteins that were often intracellular, and tended to be involved in metabolism, transcription, RNA processing or translation. In contrast, genes for secreted or plasma membrane proteins were generally expressed in only a subset of tissues. The distribution of expression levels was broad but fairly continuous: no support was found for the concept of distinct expression classes of genes. Expression estimates that included reads mapping to coding exons only correlated better with qRT-PCR data than estimates which also included 3' untranslated regions (UTRs). Muscle and liver had the least complex transcriptomes, in that they expressed predominantly ubiquitous genes and a large fraction of the transcripts came from a few highly expressed genes, whereas brain, kidney and testis expressed more complex transcriptomes with the vast majority of genes expressed and relatively small contributions from the most expressed genes. mRNAs expressed in brain had unusually long 3'UTRs, and mean 3'UTR length was higher for genes involved in development, morphogenesis and signal transduction, suggesting added complexity of UTR-based regulation for these genes. Our results support a model in which variable exterior components feed into a large, densely connected core composed of ubiquitously expressed intracellular proteins.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
-
- Bishop JO, Morton JG, Rosbash M, Richardson M. Three abundance classes in HeLa cell messenger RNA. Nature. 1974;250:199–204. - PubMed
-
- Velculescu VE, Zhang L, Vogelstein B, Kinzler KW. Serial analysis of gene expression. Science. 1995;270:484–487. - PubMed
-
- Schena M, Shalon D, Davis RW, Brown PO. Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science. 1995;270:467–470. - PubMed
-
- Lipshutz RJ, Fodor SP, Gingeras TR, Lockhart DJ. High density synthetic oligonucleotide arrays. Nat Genetics. 1999;21:20–24. - PubMed
-
- Kawai J, Shinagawa A, Shibata K, Yoshino M, Itoh M, et al. Functional annotation of a full-length mouse cDNA collection. Nature. 2001;409:685–690. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
