

Bi-directional SIFT predicts a subset of activating mutations
- PMID: 20011534
- PMCID: PMC2788704
- DOI: 10.1371/journal.pone.0008311
Bi-directional SIFT predicts a subset of activating mutations
Abstract
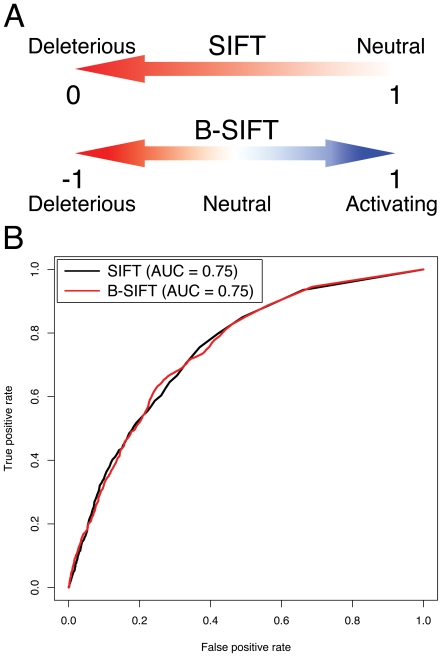
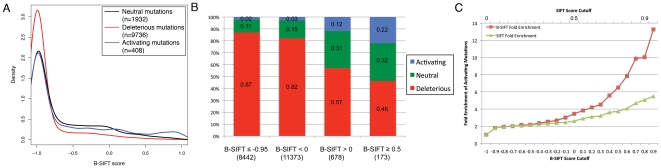
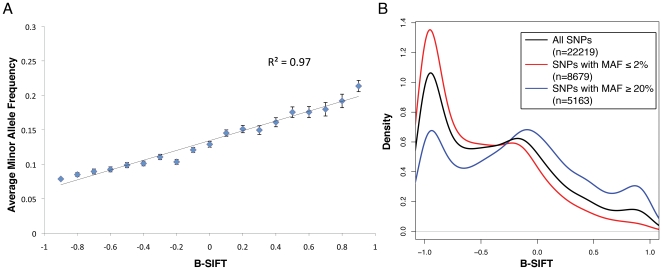
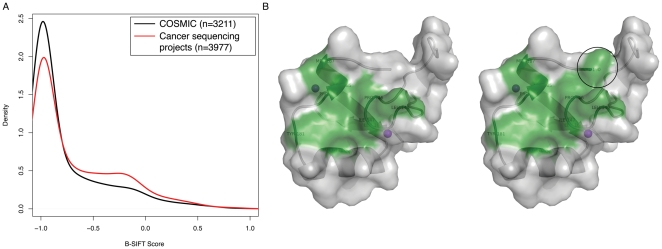
Advancements in sequencing technologies have empowered recent efforts to identify polymorphisms and mutations on a global scale. The large number of variations and mutations found in these projects requires high-throughput tools to identify those that are most likely to have an impact on function. Numerous computational tools exist for predicting which mutations are likely to be functional, but none that specifically attempt to identify mutations that result in hyperactivation or gain-of-function. Here we present a modified version of the SIFT (Sorting Intolerant from Tolerant) algorithm that utilizes protein sequence alignments with homologous sequences to identify functional mutations based on evolutionary fitness. We show that this bi-directional SIFT (B-SIFT) is capable of identifying experimentally verified activating mutants from multiple datasets. B-SIFT analysis of large-scale cancer genotyping data identified potential activating mutations, some of which we have provided detailed structural evidence to support. B-SIFT could prove to be a valuable tool for efforts in protein engineering as well as in identification of functional mutations in cancer.
Conflict of interest statement
Figures
References
-
- Karchin R, Diekhans M, Kelly L, Thomas DJ, Pieper U, et al. LS-SNP: large-scale annotation of coding non-synonymous SNPs based on multiple information sources. Bioinformatics. 2005;21:2814–2820. - PubMed
-
- Sunyaev S, Ramensky V, Koch I, Lathe W, 3rd, Kondrashov AS, et al. Prediction of deleterious human alleles. Hum Mol Genet. 2001;10:591–597. - PubMed
-
- Yue P, Li Z, Moult J. Loss of protein structure stability as a major causative factor in monogenic disease. J Mol Biol. 2005;353:459–473. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
