

Schimke immunoosseous dysplasia: defining skeletal features
- PMID: 20013129
- PMCID: PMC2876264
- DOI: 10.1007/s00431-009-1115-9
Schimke immunoosseous dysplasia: defining skeletal features
Abstract
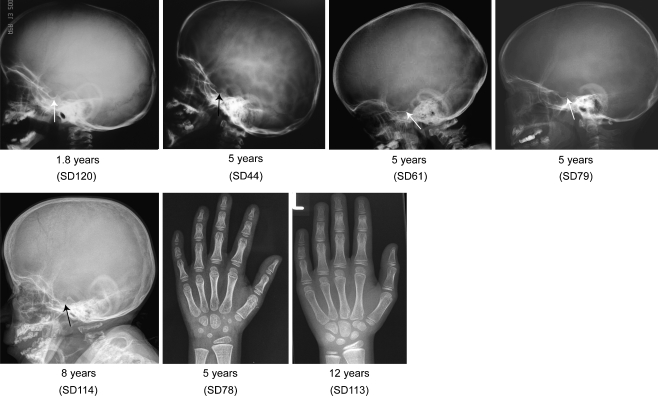
Schimke immunoosseous dysplasia (SIOD) is an autosomal recessive multisystem disorder characterized by prominent spondyloepiphyseal dysplasia, T cell deficiency, and focal segmental glomerulosclerosis. Biallelic mutations in swi/snf-related, matrix-associated, actin-dependent regulator of chromatin, subfamily a-like 1 (SMARCAL1) are the only identified cause of SIOD, but approximately half of patients referred for molecular studies do not have detectable mutations in SMARCAL1. We hypothesized that skeletal features distinguish between those with or without SMARCAL1 mutations. Therefore, we analyzed the skeletal radiographs of 22 patients with and 11 without detectable SMARCAL1 mutations. We found that patients with SMARCAL1 mutations have a spondyloepiphyseal dysplasia (SED) essentially limited to the spine, pelvis, capital femoral epiphyses, and possibly the sella turcica, whereas the hands and other long bones are basically normal. Additionally, we found that several of the adolescent and young adult patients developed osteoporosis and coxarthrosis. Of the 11 patients without detectable SMARCAL1 mutations, seven had a SED indistinguishable from patients with SMARCAL1 mutations. We conclude therefore that SED is a feature of patients with SMARCAL1 mutations and that skeletal features do not distinguish who of those with SED have SMARCAL1 mutations.
Figures





References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
