

Constitutional mismatch repair deficiency and childhood leukemia/lymphoma--report on a novel biallelic MSH6 mutation
- PMID: 20015892
- PMCID: PMC2864393
- DOI: 10.3324/haematol.2009.015503
Constitutional mismatch repair deficiency and childhood leukemia/lymphoma--report on a novel biallelic MSH6 mutation
Abstract
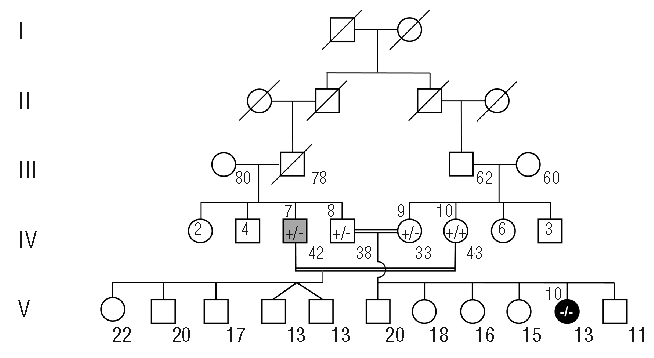
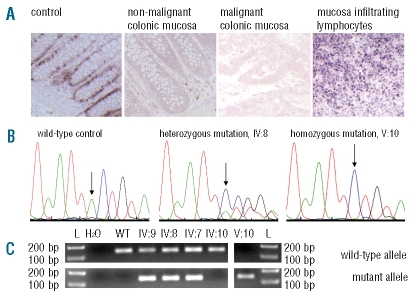
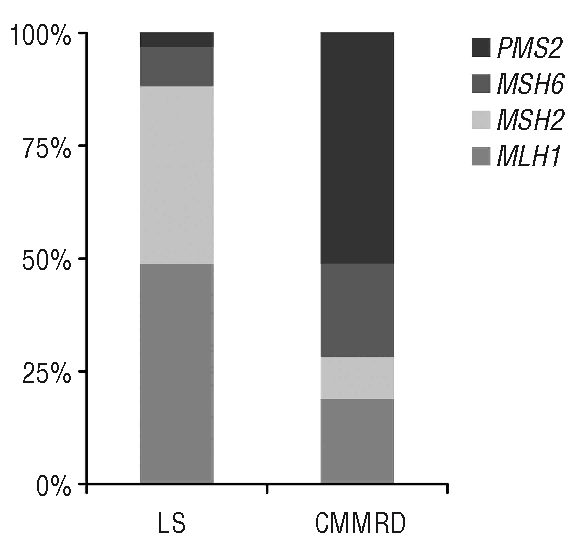
Biallelic mutations of mismatch repair genes cause constitutional mismatch repair deficiency associated with an increased risk for childhood leukemia/lymphoma. We report on a case with constitutional mismatch repair deficiency caused by a novel MSH6 mutation leading to a T-cell lymphoma and colonic adenocarcinoma at six and 13 years of age, respectively. A review of the literature on hematologic malignancies in constitutional mismatch repair deficiency showed that in almost half of the 47 known constitutional mismatch repair deficiency families, at least one individual is affected by a hematologic malignancy, predominantly T-cell lymphomas. However, diagnosing constitutional mismatch repair deficiency may be difficult when the first child is affected by leukemia/lymphoma, but identification of the causative germline mutation is of vital importance: (i) to identify relatives at risk and exclude an increased risk in non-mutation carriers; (ii) to prevent hematopoietic stem cell transplantation from sibling donors also carrying a biallelic germline mutation; and (iii) to implement effective surveillance programs for mutation carriers, that may reduce constitutional mismatch repair deficiency-associated mortality.
Figures
Comment in
-
Constitutional mismatch repair-deficiency syndrome.Haematologica. 2010 May;95(5):699-701. doi: 10.3324/haematol.2009.021626. Haematologica. 2010. PMID: 20442441 Free PMC article. No abstract available.
References
-
- Jiricny J. The multifaceted mismatch-repair system. Nat Rev Mol Cell Biol. 2006;7(5):335–46. - PubMed
-
- Lynch HT, de la Chapelle A. Hereditary colorectal cancer. N Engl J Med. 2003;348(10):919–32. - PubMed
-
- Lynch HT, Lynch JF, Lynch PM, Attard T. Hereditary colorectal cancer syndromes: molecular genetics, genetic counseling, diagnosis and management. Fam Cancer. 2008;7(1):27–39. - PubMed
-
- Scott RH, Mansour S, Pritchard-Jones K, Kumar D, MacSweeney F, Rahman N. Medulloblastoma, acute myelocytic leukemia and colonic carcinomas in a child with biallelic MSH6 mutations. Nat Clin Pract Oncol. 2007;4(2):130–4. - PubMed
-
- Wimmer K, Etzler J. Constitutional mismatch repair-deficiency syndrome: have we so far seen only the tip of an iceberg? Hum Genet. 2008;124(2):105–22. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
