

Complementary Tolls in the periodontium: how periodontal bacteria modify complement and Toll-like receptor responses to prevail in the host
- PMID: 20017800
- PMCID: PMC2796596
- DOI: 10.1111/j.1600-0757.2009.00324.x
Complementary Tolls in the periodontium: how periodontal bacteria modify complement and Toll-like receptor responses to prevail in the host
Abstract
The complement and the Toll-like receptors are rapidly activatable systems which, in concert, provide first-line innate defense against infection and act as mediators between the innate and the adaptive immune response. The ability of periodontal bacteria to persist and establish chronic infections in the periodontium suggests that they may have evolved strategies to evade, disarm, or subvert these defense systems to their own advantage. Indeed, accumulating evidence indicates that at least some of the major periodontal pathogens utilize ingenious mechanisms to not only undermine each system separately, but also exploit crosstalk points between the complement and the Toll-like receptor pathways. It is conceivable that immune subversive activities by certain keynote periodontal pathogens, such as those comprising the so-called “red complex”, may be critical for the persistence of the entire mixed-species biofilm community in the diseased periodontium. This review summarizes and synthesizes recent discoveries in this field, which offers important insights into the pathology associated with the complex periodontal host-microbe interplay.
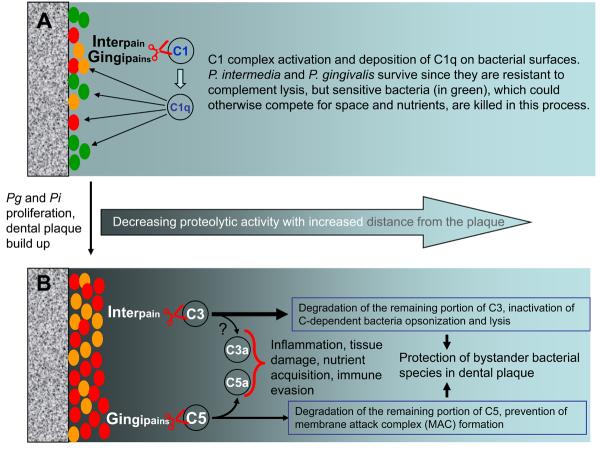
Figures
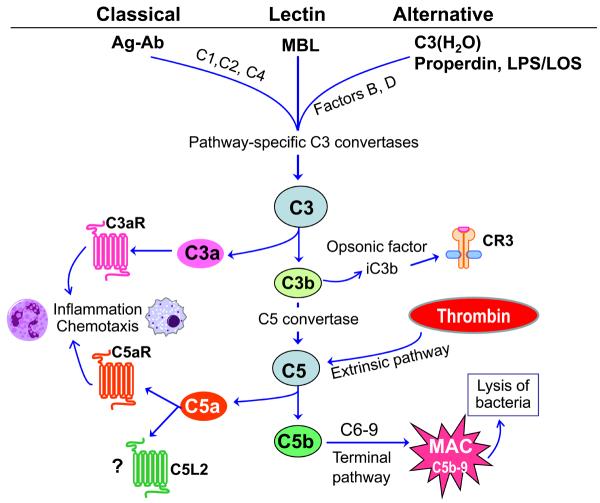
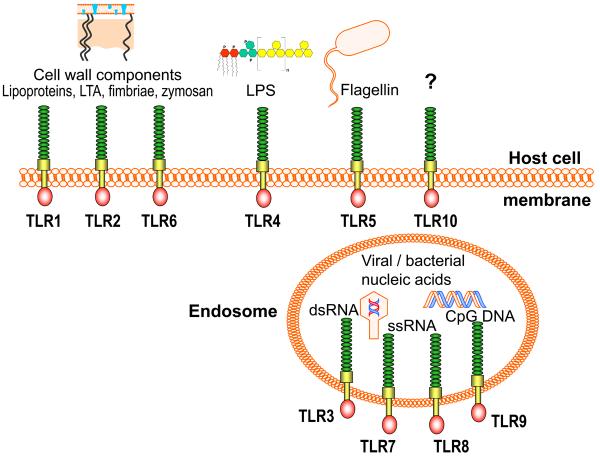
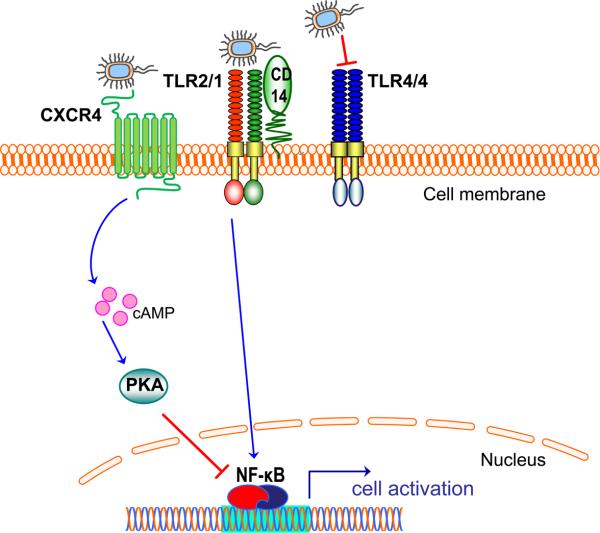
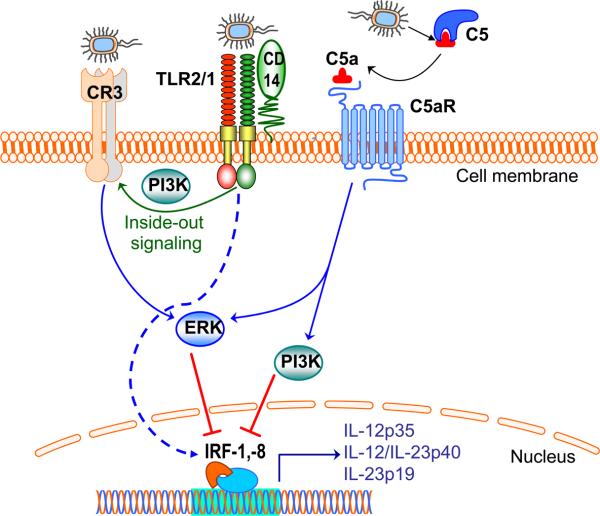
References
-
- Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol. 2004;4:499–511. - PubMed
-
- Alexander JJ, Quigg RJ. The simple design of complement factor H: Looks can be deceiving. Mol Immunol. 2007;44:123–132. - PubMed
-
- Andreakos E, Foxwell B, Feldmann M. Is targeting Toll-like receptors and their signaling pathway a useful therapeutic approach to modulating cytokine-driven inflammation? Immunol Rev. 2004;202:250–265. - PubMed
-
- Arbibe L, Mira JP, Teusch N, Kline L, Guha M, Mackman N, Godowski PJ, Ulevitch RJ, Knaus UG. Toll-like receptor 2-mediated NF-κB activation requires a Rac1-dependent pathway. Nat Immunol. 2000;1:533–540. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R01 DE009761/DE/NIDCR NIH HHS/United States
- R01 DE015254/DE/NIDCR NIH HHS/United States
- AI068730/AI/NIAID NIH HHS/United States
- DE017138/DE/NIDCR NIH HHS/United States
- AI072106/AI/NIAID NIH HHS/United States
- DE018292/DE/NIDCR NIH HHS/United States
- GM062134/GM/NIGMS NIH HHS/United States
- R01 GM062134/GM/NIGMS NIH HHS/United States
- N01 AI030040/AI/NIAID NIH HHS/United States
- R01 DE017138/DE/NIDCR NIH HHS/United States
- R01 DE018292/DE/NIDCR NIH HHS/United States
- R01 AI030040/AI/NIAID NIH HHS/United States
- DE009761/DE/NIDCR NIH HHS/United States
- DE015254/DE/NIDCR NIH HHS/United States
- P01 AI068730/AI/NIAID NIH HHS/United States
- R01 AI072106/AI/NIAID NIH HHS/United States
LinkOut - more resources
Full Text Sources
