

Transforming growth factor beta (TGF-beta) and inflammation in cancer
- PMID: 20018551
- PMCID: PMC2834863
- DOI: 10.1016/j.cytogfr.2009.11.008
Transforming growth factor beta (TGF-beta) and inflammation in cancer
Abstract
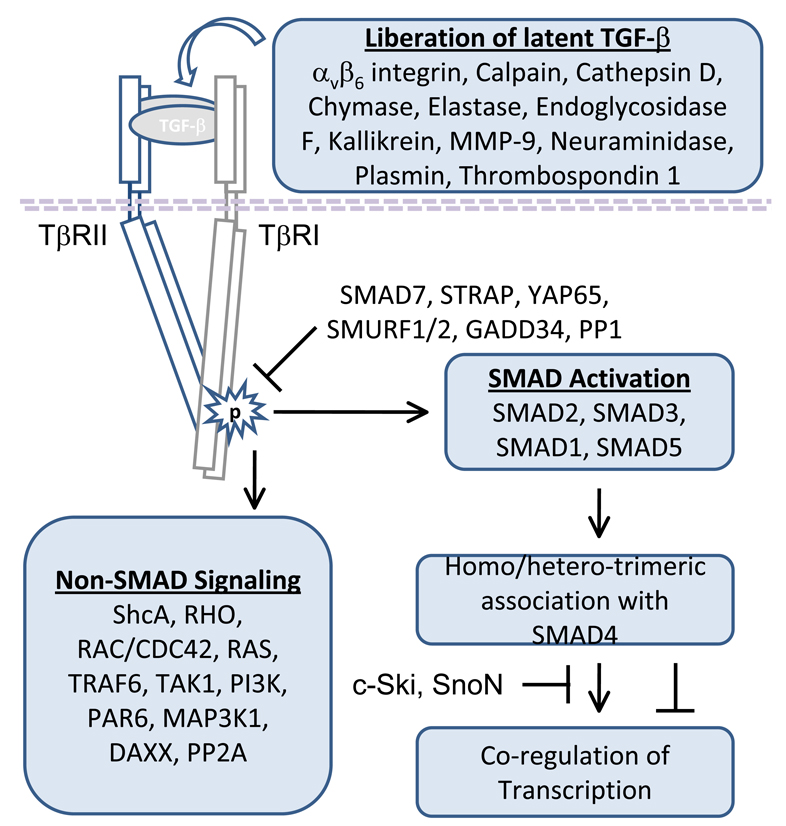
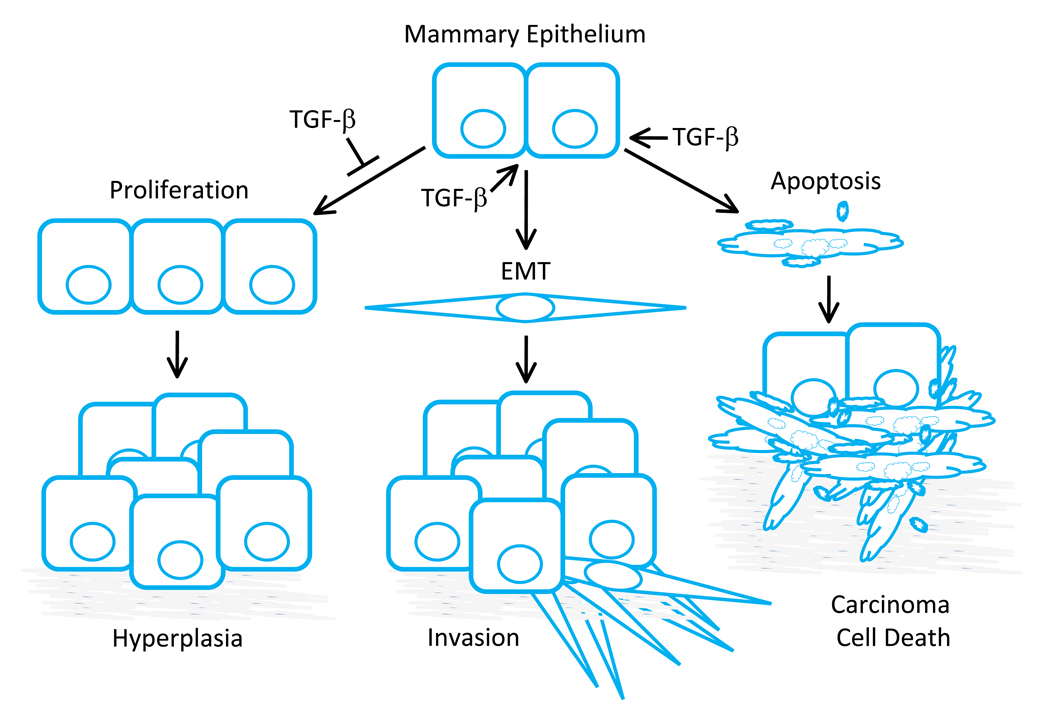
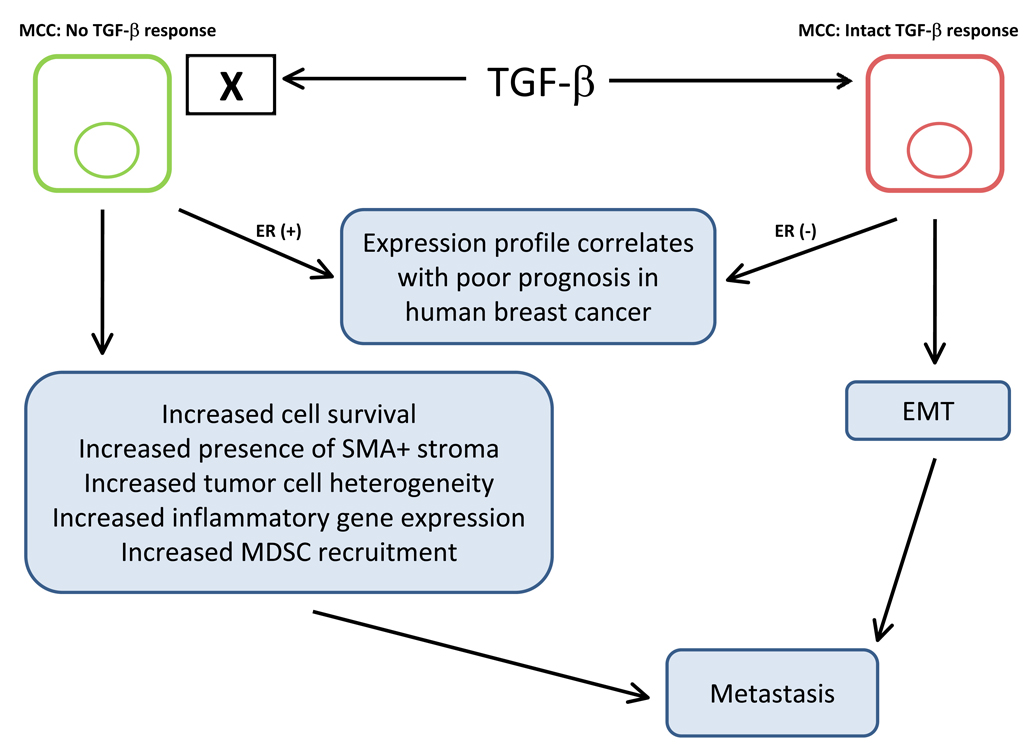
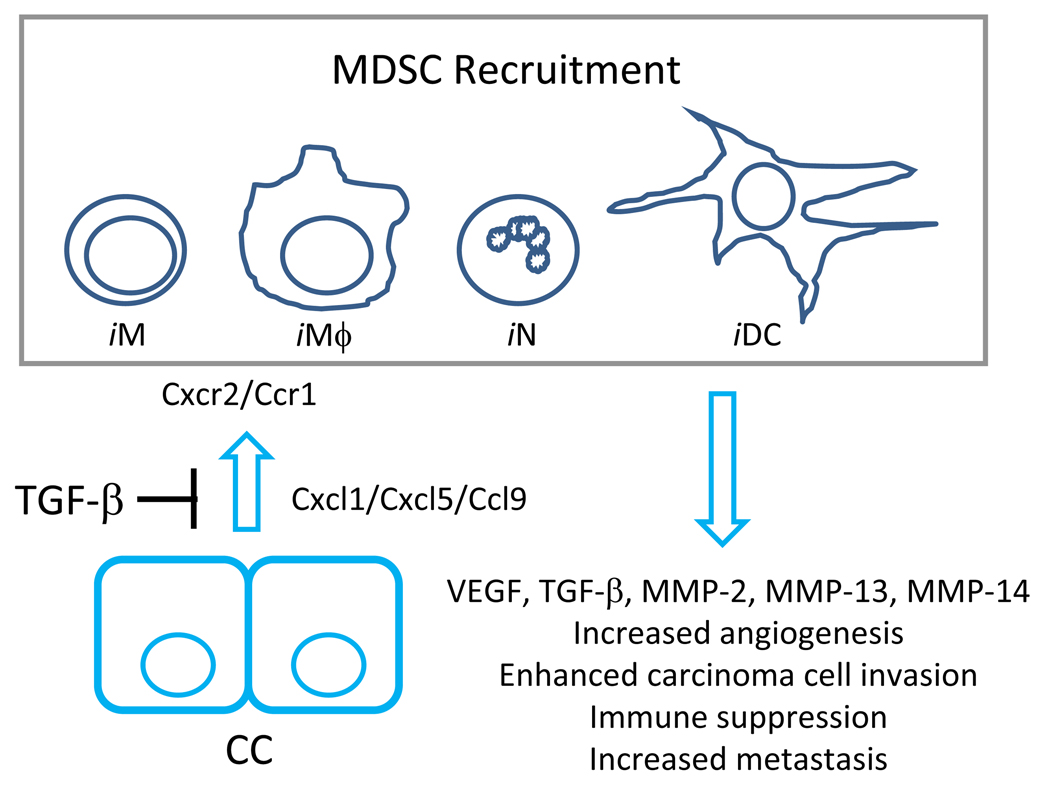
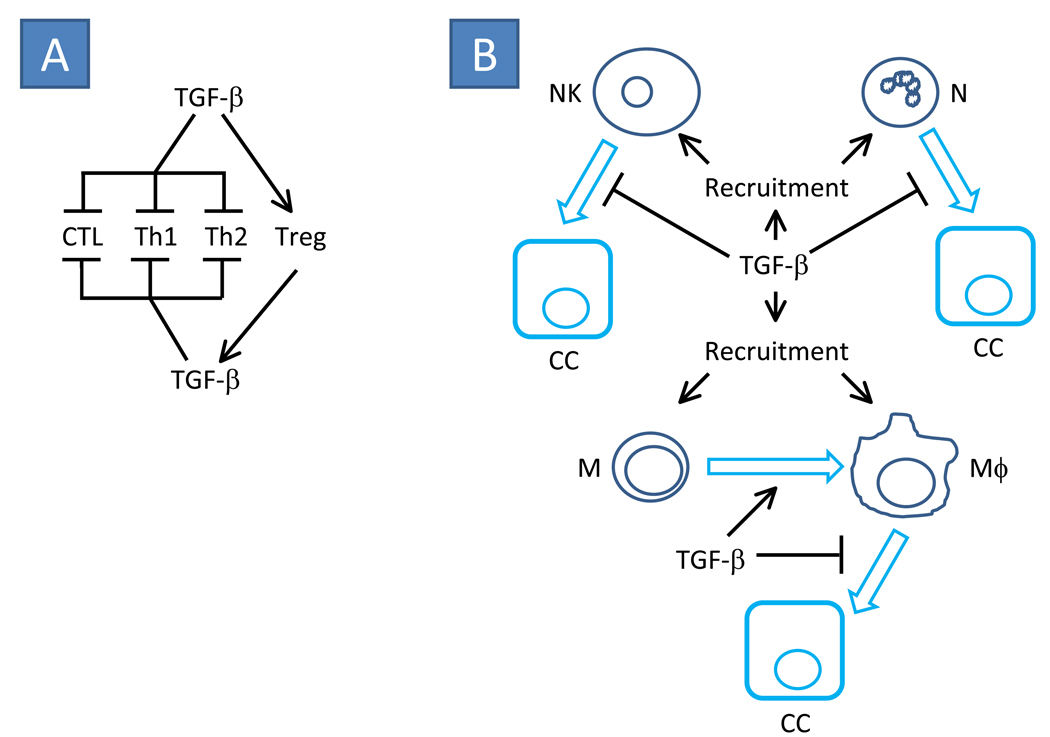
The transforming growth factor beta (TGF-beta) has been studied with regard to the regulation of cell behavior for over three decades. A large body of research has been devoted to the regulation of epithelial cell and derivative carcinoma cell populations in vitro and in vivo. TGF-beta has been shown to inhibit epithelial cell cycle progression and promote apoptosis that together significantly contribute to the tumor suppressive role for TGF-beta during carcinoma initiation and progression. TGF-beta is also able to promote an epithelial to mesenchymal transition that has been associated with increased tumor cell motility, invasion and metastasis. However, it has now been shown that loss of carcinoma cell responsiveness to TGF-beta stimulation can also promote metastasis. Interestingly, enhanced metastasis in the absence of a carcinoma cell response to TGF-beta stimulation has been shown to involve increased chemokine production resulting in recruitment of pro-metastatic myeloid derived suppressor cell (MDSC) populations to the tumor microenvironment at the leading invasive edge. When present, MDSCs enhance angiogenesis, promote immune tolerance and provide matrix degrading enzymes that promote tumor progression and metastasis. Further, the recruitment of MDSC populations in this context likely enhances the classic role for TGF-beta in immune suppression since the MDSCs are an abundant source of TGF-beta production. Importantly, it is now clear that carcinoma-immune cell cross-talk initiated by TGF-beta signaling within the carcinoma cell is a significant determinant worth consideration when designing therapeutic strategies to manage tumor progression and metastasis.
Figures
References
-
- Kehrl JH, et al. Transforming growth factor beta is an important immunomodulatory protein for human B lymphocytes. J Immunol. 1986;137(12):3855–3860. - PubMed
-
- Rook AH, et al. Effects of transforming growth factor beta on the functions of natural killer cells: depressed cytolytic activity and blunting of interferon responsiveness. J Immunol. 1986;136(10):3916–3920. - PubMed
-
- Fontana A, et al. Expression of TGF-beta 2 in human glioblastoma: a role in resistance to immune rejection? Ciba Found Symp. 1991;157:232–238. discussion 238-41. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
