

Homozygous frameshift mutation in TMCO1 causes a syndrome with craniofacial dysmorphism, skeletal anomalies, and mental retardation
- PMID: 20018682
- PMCID: PMC2806776
- DOI: 10.1073/pnas.0908457107
Homozygous frameshift mutation in TMCO1 causes a syndrome with craniofacial dysmorphism, skeletal anomalies, and mental retardation
Abstract
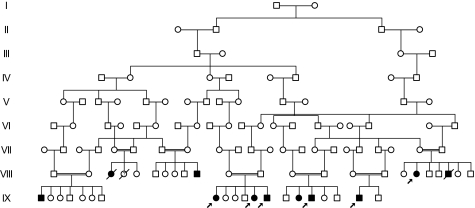
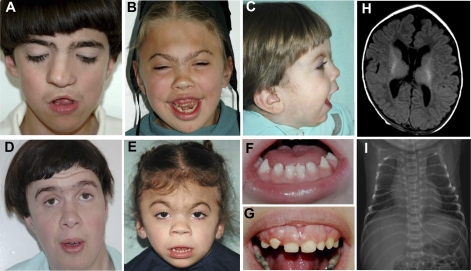
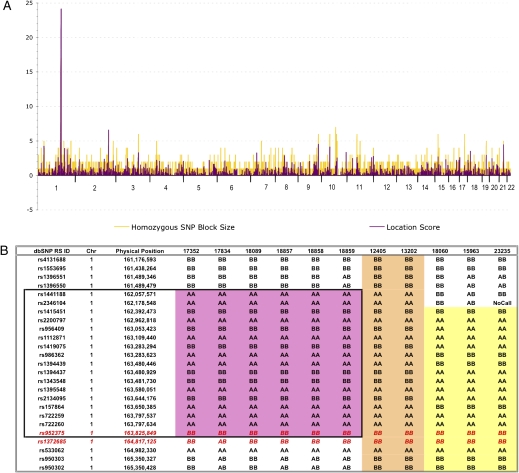
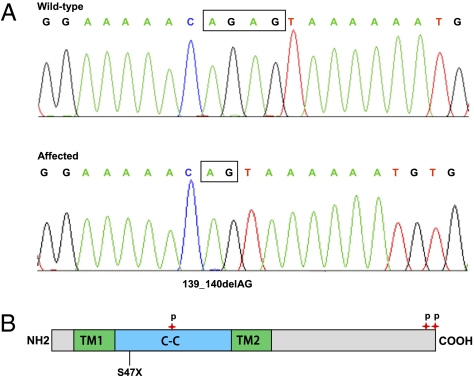
We identified an autosomal recessive condition in 11 individuals in the Old Order Amish of northeastern Ohio. The syndrome was characterized by distinctive craniofacial dysmorphism, skeletal anomalies, and mental retardation. The typical craniofacial dysmorphism included brachycephaly, highly arched bushy eyebrows, synophrys, long eyelashes, low-set ears, microdontism of primary teeth, and generalized gingival hyperplasia, whereas Sprengel deformity of scapula, fusion of spine, rib abnormities, pectus excavatum, and pes planus represented skeletal anomalies. The genome-wide homozygosity mapping using six affected individuals localized the disease gene to a 3.3-Mb region on chromosome 1q23.3-q24.1. Candidate gene sequencing identified a homozygous frameshift mutation, c.139_140delAG, in the transmembrane and coiled-coil domains 1 (TMCO1) gene, as the pathogenic change in all affected members of the extended pedigree. This mutation is predicted to result in a severely truncated protein (p.Ser47Ter) of only one-fourth the original length. The TMCO1 gene product is a member of DUF841 superfamily of several eukaryotic proteins with unknown function. The gene has highly conserved amino acid sequence and is universally expressed in all human tissues examined. The high degree of conservation and the ubiquitous expression pattern in human adult and fetal tissues suggest a critical role for TMCO1. This report shows a TMCO1 sequence variant being associated with a genetic disorder in human. We propose "TMCO1 defect syndrome" as the name of this condition.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Craniofacial dysmorphism, skeletal anomalies, and impaired intellectual development syndrome-1 in two new patients with the same homozygous TMCO1 variant and review of the literature.Eur J Med Genet. 2023 Mar;66(3):104715. doi: 10.1016/j.ejmg.2023.104715. Epub 2023 Jan 25. Eur J Med Genet. 2023. PMID: 36708876 Review.
-
TMCO1 deficiency causes autosomal recessive cerebrofaciothoracic dysplasia.Am J Med Genet A. 2014 Feb;164A(2):291-304. doi: 10.1002/ajmg.a.36248. Epub 2013 Nov 5. Am J Med Genet A. 2014. PMID: 24194475 Review.
-
Cerebrofaciothoracic dysplasia: Four new patients with a recurrent TMCO1 pathogenic variant.Am J Med Genet A. 2019 Jan;179(1):43-49. doi: 10.1002/ajmg.a.60678. Epub 2018 Dec 17. Am J Med Genet A. 2019. PMID: 30556256
-
Whole-exome sequencing links TMCO1 defect syndrome with cerebro-facio-thoracic dysplasia.Eur J Hum Genet. 2014 Sep;22(9):1145-8. doi: 10.1038/ejhg.2013.291. Epub 2014 Jan 15. Eur J Hum Genet. 2014. PMID: 24424126 Free PMC article.
-
A novel biallelic loss-of-function mutation in TMCO1 gene confirming and expanding the phenotype spectrum of cerebro-facio-thoracic dysplasia.Am J Med Genet A. 2019 Jul;179(7):1338-1345. doi: 10.1002/ajmg.a.61168. Epub 2019 May 18. Am J Med Genet A. 2019. PMID: 31102500
Cited by
-
Substrate-driven assembly of a translocon for multipass membrane proteins.Nature. 2022 Nov;611(7934):167-172. doi: 10.1038/s41586-022-05330-8. Epub 2022 Oct 19. Nature. 2022. PMID: 36261522 Free PMC article.
-
Homozygous mutation in SAMHD1 gene causes cerebral vasculopathy and early onset stroke.Proc Natl Acad Sci U S A. 2011 Mar 29;108(13):5372-7. doi: 10.1073/pnas.1014265108. Epub 2011 Mar 14. Proc Natl Acad Sci U S A. 2011. PMID: 21402907 Free PMC article.
-
Common genetic determinants of intraocular pressure and primary open-angle glaucoma.PLoS Genet. 2012;8(5):e1002611. doi: 10.1371/journal.pgen.1002611. Epub 2012 May 3. PLoS Genet. 2012. PMID: 22570627 Free PMC article.
-
Genome-wide association study identifies susceptibility loci for open angle glaucoma at TMCO1 and CDKN2B-AS1.Nat Genet. 2011 Jun;43(6):574-8. doi: 10.1038/ng.824. Epub 2011 May 1. Nat Genet. 2011. PMID: 21532571
-
Cerebro-facio-thoracic dysplasia (Pascual-Castroviejo syndrome): Identification of a novel mutation, use of facial recognition analysis, and review of the literature.Transl Sci Rare Dis. 2018 Apr 13;3(1):37-43. doi: 10.3233/TRD-180022. Transl Sci Rare Dis. 2018. PMID: 29682451 Free PMC article.
References
-
- Raymond FL, Tarpey P. The genetics of mental retardation. Hum Mol Genet. 2006;15(Spec No 2):R110–R116. - PubMed
-
- Strauss KA, et al. Recessive symptomatic focal epilepsy and mutant contactin-associated protein-like 2. N Engl J Med. 2006;354:1370–1377. - PubMed
-
- Xin B, Puffenberger EG, Tumbush J, Bockoven JR, Wang H. Homozygosity for a novel splice site mutation in the cardiac myosin-binding protein C gene causes severe neonatal hypertrophic cardiomyopathy. Am J Med Genet A. 2007;143:2662–2667. - PubMed
-
- Xin B, Puffenberger EG, Nye L, Wiznitzer M, Wang H. A novel mutation in the GDAP1 gene is associated with autosomal recessive Charcot-Marie-Tooth disease in an Amish family. Clin Genet. 2008;74:274–278. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
