

High-resolution protein complexes from integrating genomic information with molecular simulation
- PMID: 20018738
- PMCID: PMC2799721
- DOI: 10.1073/pnas.0912100106
High-resolution protein complexes from integrating genomic information with molecular simulation
Abstract
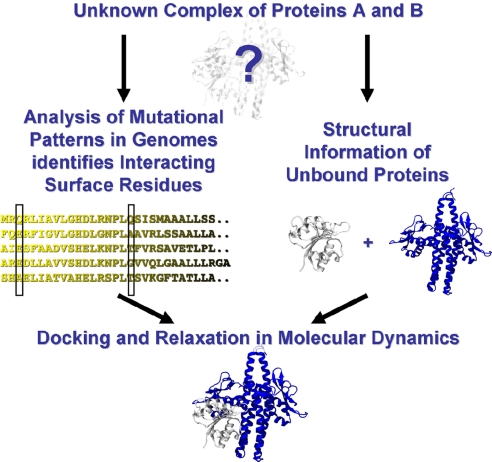
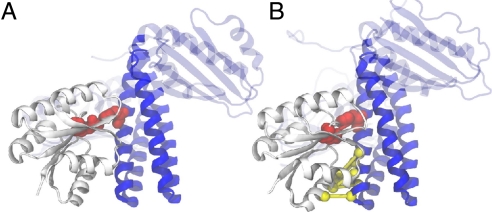
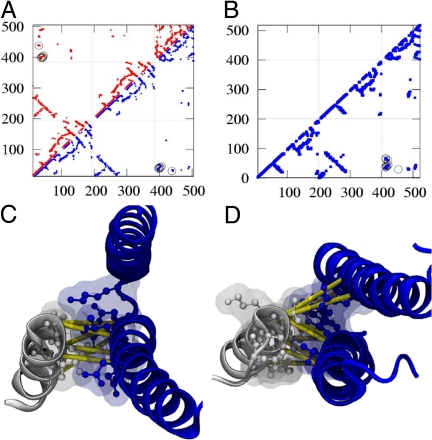
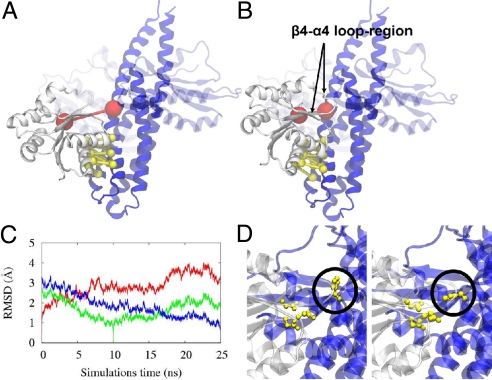
Bacteria use two-component signal transduction systems (TCS) extensively to sense and react to external stimuli. In these, a membrane-bound sensor histidine kinase (SK) autophosphorylates in response to an environmental stimulus and transfers the phosphoryl group to a transcription factor/response regulator (RR) that mediates the cellular response. The complex between these two proteins is ruled by transient interactions, which provides a challenge to experimental structure determination techniques. The functional and structural homolog of an SK/RR pair Spo0B/Spo0F, however, has been structurally resolved. Here, we describe a method capable of generating structural models of such transient protein complexes. By using existing structures of the individual proteins, our method combines bioinformatically derived contact residue information with molecular dynamics simulations. We find crystal resolution accuracy with existing crystallographic data when reconstituting the known system Spo0B/Spo0F. Using this approach, we introduce a complex structure of TM0853/TM0468 as an exemplary SK/RR TCS, consistent with all experimentally available data.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
The signaling pathway in histidine kinase and the response regulator complex revealed by X-ray crystallography and solution scattering.J Mol Biol. 2006 Sep 8;362(1):123-39. doi: 10.1016/j.jmb.2006.07.012. Epub 2006 Jul 15. J Mol Biol. 2006. PMID: 16890956
-
Computational modeling of phosphotransfer complexes in two-component signaling.Methods Enzymol. 2010;471:43-58. doi: 10.1016/S0076-6879(10)71003-X. Epub 2010 Mar 1. Methods Enzymol. 2010. PMID: 20946841
-
Structure of PAS-linked histidine kinase and the response regulator complex.Structure. 2009 Oct 14;17(10):1333-44. doi: 10.1016/j.str.2009.07.016. Structure. 2009. PMID: 19836334
-
Bacterial histidine kinase as signal sensor and transducer.Int J Biochem Cell Biol. 2006 Mar;38(3):307-12. doi: 10.1016/j.biocel.2005.08.018. Epub 2005 Sep 26. Int J Biochem Cell Biol. 2006. PMID: 16242988 Review.
-
Structural basis of the signal transduction in the two-component system.Adv Exp Med Biol. 2008;631:22-39. doi: 10.1007/978-0-387-78885-2_3. Adv Exp Med Biol. 2008. PMID: 18792680 Review.
Cited by
-
Design of a Protein with Improved Thermal Stability by an Evolution-Based Generative Model.Angew Chem Int Ed Engl. 2022 Dec 12;61(50):e202202711. doi: 10.1002/anie.202202711. Epub 2022 Nov 16. Angew Chem Int Ed Engl. 2022. PMID: 36259321 Free PMC article.
-
Biophysics of protein evolution and evolutionary protein biophysics.J R Soc Interface. 2014 Nov 6;11(100):20140419. doi: 10.1098/rsif.2014.0419. J R Soc Interface. 2014. PMID: 25165599 Free PMC article.
-
Native structure-based modeling and simulation of biomolecular systems per mouse click.BMC Bioinformatics. 2014 Aug 29;15(1):292. doi: 10.1186/1471-2105-15-292. BMC Bioinformatics. 2014. PMID: 25176255 Free PMC article.
-
Dimeric interactions and complex formation using direct coevolutionary couplings.Sci Rep. 2015 Sep 4;5:13652. doi: 10.1038/srep13652. Sci Rep. 2015. PMID: 26338201 Free PMC article.
-
Co-Evolutionary Fitness Landscapes for Sequence Design.Angew Chem Int Ed Engl. 2018 May 14;57(20):5674-5678. doi: 10.1002/anie.201713220. Epub 2018 Mar 25. Angew Chem Int Ed Engl. 2018. PMID: 29512300 Free PMC article.
References
-
- Wells JA, McClendon CL. Reaching for high-hanging fruit in drug discovery at protein–protein interfaces. Nature. 2007;450:1001–1009. - PubMed
-
- Eswar N, Eramian D, Webb B, Shen M-Y, Sali A. Methods in Molecular Biology. Totowa, NJ: Humana; 2008. pp. 145–159. - PubMed
-
- Fujitsuka Y, Takada S, Luthey-Schulten ZA, Wolynes PG. Optimizing physical energy functions for protein folding. Proteins. 2004;54:88–103. - PubMed
-
- Hansmann UHE, Okamoto Y. Prediction of peptide conformations by multicanonical algorithm—New approach to the multiple-minima problem. J Comput Chem. 1993;14:1333–1338.
-
- Schug A, Herges T, Wenzel W. Reproducible protein folding with the stochastic tunneling method. Phys Rev Lett. 2003;91:158102. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
