

The neuropeptide calcitonin gene-related peptide causes repression of tumor necrosis factor-alpha transcription and suppression of ATF-2 promoter recruitment in Toll-like receptor-stimulated dendritic cells
- PMID: 20018859
- PMCID: PMC2823491
- DOI: 10.1074/jbc.M109.066787
The neuropeptide calcitonin gene-related peptide causes repression of tumor necrosis factor-alpha transcription and suppression of ATF-2 promoter recruitment in Toll-like receptor-stimulated dendritic cells
Abstract
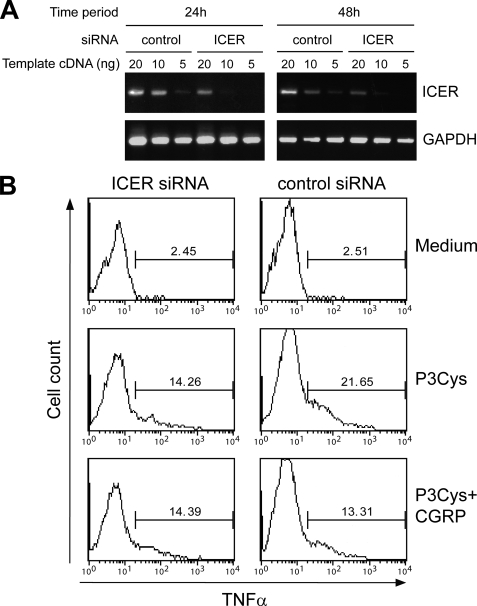
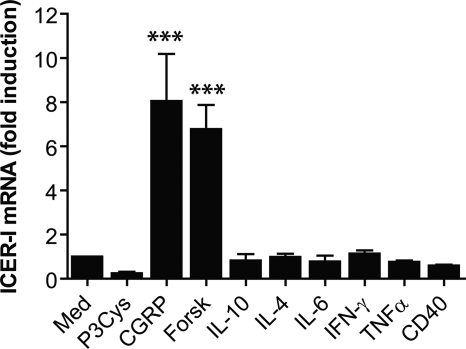
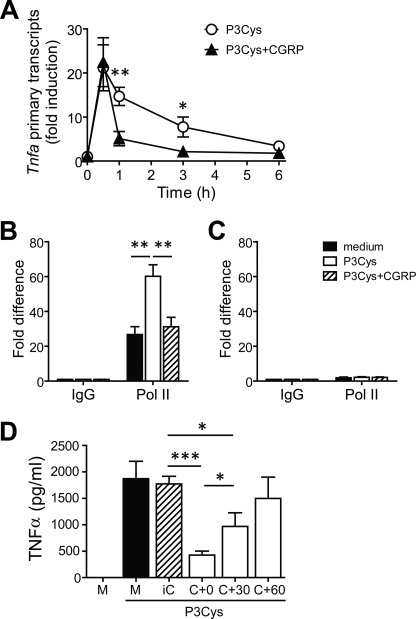
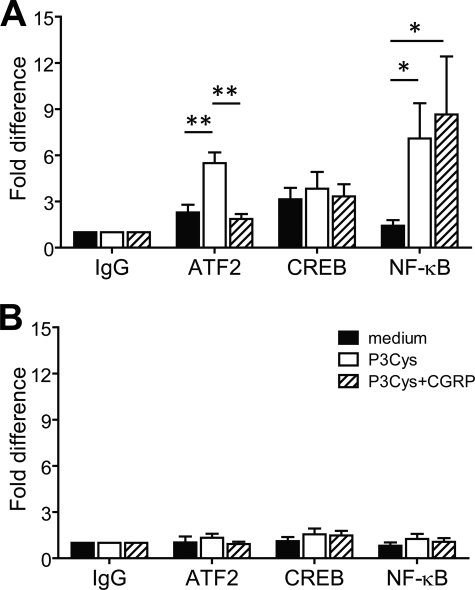
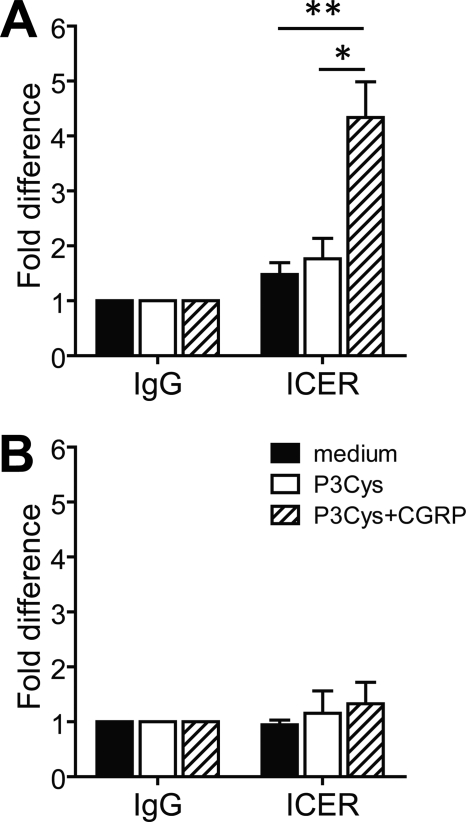
Sensory nerves may dampen inflammatory processes through the release of the neuropeptide calcitonin gene-related peptide (CGRP). CGRP mediates immunosuppressive activities through up-regulation of interleukin-10 or, alternatively, through an interleukin-10-independent pathway that is associated with rapid induction of the transcriptional inducible cAMP early repressor (ICER). In this work, we further investigated the molecular mechanisms of immune modulation by CGRP. Using TLR2-stimulated dendritic cells, we show that inhibition of tumor necrosis factor-alpha production by CGRP is dependent on up-regulation of endogenous ICER. Dendritic cell expression of ICER was selectively induced by CGRP and elevation of cellular cAMP levels but not by numerous pro- and anti-inflammatory cytokines. Treatment of dendritic cells with CGRP did not interfere with the induction of Tnfa gene expression but caused premature repression of TLR2-induced transcriptional activity. ATF-2 was rapidly phosphorylated and recruited to the Tnfa promoter following ligation of TLR2. Concomitant administration of CGRP completely prevented binding of ATF-2 to the Tnfa promoter, whereas recruitment of ICER was markedly elevated. In contrast, CGRP did not influence TLR2-stimulated binding of NF-kappaB p65. Together, these results are consistent with a model suggesting that CGRP causes rapid up-regulation of ICER, which in turn competes with ATF-2 for binding to the Tnfa promoter, leading to repression of gene expression.
Figures
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
