

A robust error model for iTRAQ quantification reveals divergent signaling between oncogenic FLT3 mutants in acute myeloid leukemia
- PMID: 20019052
- PMCID: PMC2871413
- DOI: 10.1074/mcp.M900452-MCP200
A robust error model for iTRAQ quantification reveals divergent signaling between oncogenic FLT3 mutants in acute myeloid leukemia
Abstract
The FLT3 receptor tyrosine kinase plays an important role in normal hematopoietic development and leukemogenesis. Point mutations within the activation loop and in-frame tandem duplications of the juxtamembrane domain represent the most frequent molecular abnormalities observed in acute myeloid leukemia. Interestingly these gain-of-function mutations correlate with different clinical outcomes, suggesting that signals from constitutive FLT3 mutants activate different downstream targets. In principle, mass spectrometry offers a powerful means to quantify protein phosphorylation and identify signaling events associated with constitutively active kinases or other oncogenic events. However, regulation of individual phosphorylation sites presents a challenging case for proteomics studies whereby quantification is based on individual peptides rather than an average across different peptides derived from the same protein. Here we describe a robust experimental framework and associated error model for iTRAQ-based quantification on an Orbitrap mass spectrometer that relates variance of peptide ratios to mass spectral peak height and provides for assignment of p value, q value, and confidence interval to every peptide identification, all based on routine measurements, obviating the need for detailed characterization of individual ion peaks. Moreover, we demonstrate that our model is stable over time and can be applied in a manner directly analogous to ubiquitously used external mass calibration routines. Application of our error model to quantitative proteomics data for FLT3 signaling provides evidence that phosphorylation of tyrosine phosphatase SHP1 abrogates the transformative potential, but not overall kinase activity, of FLT3-D835Y in acute myeloid leukemia.
Figures
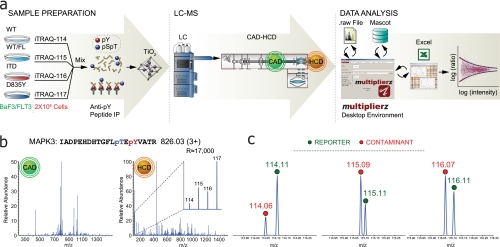
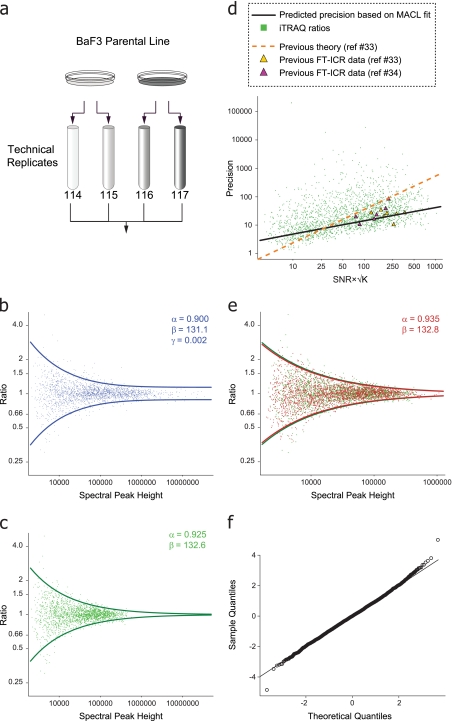
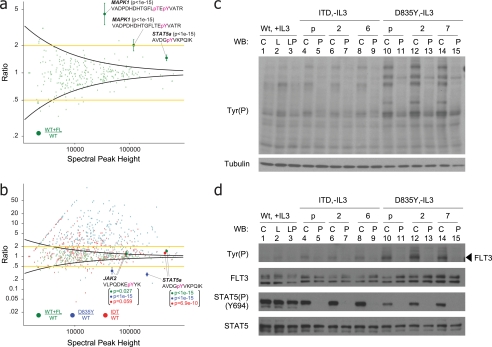
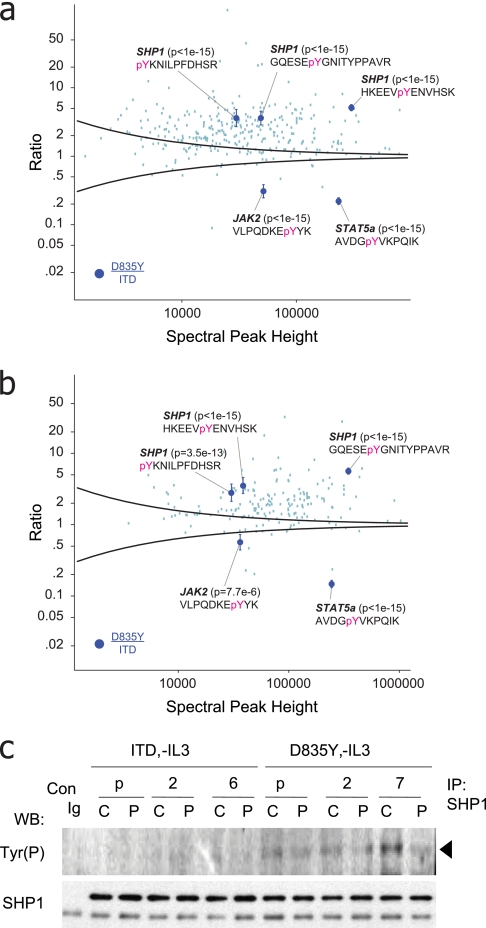
References
-
- Blume-Jensen P., Hunter T. (2001) Oncogenic kinase signalling. Nature 411, 355–365 - PubMed
-
- Schlessinger J. (2000) Cell signaling by receptor tyrosine kinases. Cell 103, 211–225 - PubMed
-
- Gilliland D. G., Griffin J. D. (2002) The roles of FLT3 in hematopoiesis and leukemia. Blood 100, 1532–1542 - PubMed
-
- Small D. (2006) FLT3 mutations: biology and treatment. Hematology Am. Soc. Hematol. Educ. Program 1, 178–184 - PubMed
-
- Knapper S. (2007) FLT3 inhibition in acute myeloid leukaemia. Br. J. Haematol 138, 687–699 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
