

Glucose restriction can extend normal cell lifespan and impair precancerous cell growth through epigenetic control of hTERT and p16 expression
- PMID: 20019239
- PMCID: PMC2996891
- DOI: 10.1096/fj.09-149328
Glucose restriction can extend normal cell lifespan and impair precancerous cell growth through epigenetic control of hTERT and p16 expression
Erratum in
-
Correction to "Glucose Restriction Can Extend Normal Cell Lifespan and Impair Precancerous Cell Growth Through Epigenetic Control of hTERT and p16 Expression".FASEB J. 2025 Jun 30;39(12):e70731. doi: 10.1096/fj.202501897. FASEB J. 2025. PMID: 40540356 No abstract available.
Abstract
Cancer cells metabolize glucose at elevated rates and have a higher sensitivity to glucose reduction. However, the precise molecular mechanisms leading to different responses to glucose restriction between normal and cancer cells are not fully understood. We analyzed normal WI-38 and immortalized WI-38/S fetal lung fibroblasts and found that glucose restriction resulted in growth inhibition and apoptosis in WI-38/S cells, whereas it induced lifespan extension in WI-38 cells. Moreover, in WI-38/S cells glucose restriction decreased expression of hTERT (human telomerase reverse transcriptase) and increased expression of p16(INK4a). Opposite effects were found in the gene expression of hTERT and p16 in WI-38 cells in response to glucose restriction. The altered gene expression was partly due to glucose restriction-induced DNA methylation changes and chromatin remodeling of the hTERT and p16 promoters in normal and immortalized WI-38 cells. Furthermore, glucose restriction resulted in altered hTERT and p16 expression in response to epigenetic regulators in WI-38 rather than WI-38/S cells, suggesting that energy stress-induced differential epigenetic regulation may lead to different cellular fates in normal and precancerous cells. Collectively, these results provide new insights into the epigenetic mechanisms of a nutrient control strategy that may contribute to cancer therapy as well as antiaging approaches.
Figures





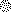 , partial methylated cytosine. C) Sequencing at ∼−190 of the p16 promoter, which is the putative E2F-1 binding site. Arrows indicate changed CpG sites of the E2F-1 binding site. D) Chromatin DNA from glucose restriction-treated and untreated WI-38 cells was immunoprecipitated by E2F-1 antibody together with mouse IgG controls. Photograph is representative of an experiment that was repeated in triplicate. E) Schematic presentation of E2F-1 blocking access to the methylated E2F-1 binding site in the p16 promoter in response to glucose restriction.
, partial methylated cytosine. C) Sequencing at ∼−190 of the p16 promoter, which is the putative E2F-1 binding site. Arrows indicate changed CpG sites of the E2F-1 binding site. D) Chromatin DNA from glucose restriction-treated and untreated WI-38 cells was immunoprecipitated by E2F-1 antibody together with mouse IgG controls. Photograph is representative of an experiment that was repeated in triplicate. E) Schematic presentation of E2F-1 blocking access to the methylated E2F-1 binding site in the p16 promoter in response to glucose restriction.

References
-
- Thompson C., Bauer D., Lum J., Hatzivassiliou G., Zong W., Zhao F., Ditsworth D., Buzzai M., Lindsten T. How do cancer cells acquire the fuel needed to support cell growth? Cold Spring Harb Symp Quant Biol. 2005;70:357–362. - PubMed
-
- Garber K. Energy deregulation: licensing tumors to grow. Science. 2006;312:1158–1159. - PubMed
-
- Zhu Z., Jiang W., McGinley J., Price J., Gao B., Thompson H. Effects of dietary energy restriction on gene regulation in mammary epithelial cells. Cancer Res. 2007;67:12018–12025. - PubMed
-
- Warburg O. On the origin of cancer cells. Science. 1956;123:309–314. - PubMed
-
- Lunt S., Chaudary N., Hill R. The tumor microenvironment and metastatic disease. Clin Exp Metastasis. 2009;26:19–34. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
