

CCL2 is a negative regulator of AMP-activated protein kinase to sustain mTOR complex-1 activation, survivin expression, and cell survival in human prostate cancer PC3 cells
- PMID: 20019839
- PMCID: PMC2794512
- DOI: 10.1593/neo.09936
CCL2 is a negative regulator of AMP-activated protein kinase to sustain mTOR complex-1 activation, survivin expression, and cell survival in human prostate cancer PC3 cells
Abstract
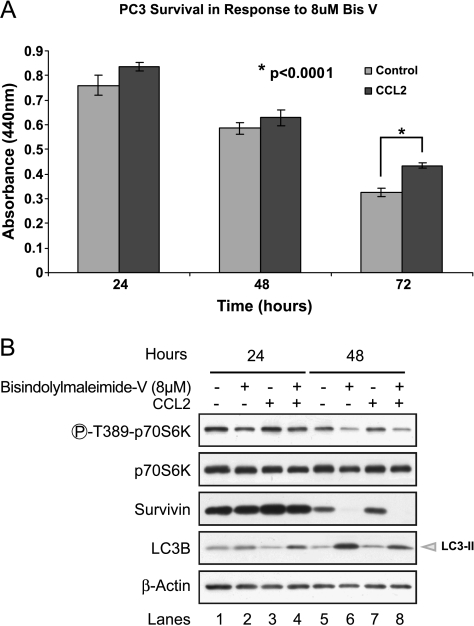
CCL2 is a cytokine prevalent in the prostate cancer tumor microenvironment. Recently, we reported that CCL2 induces the mammalian target of rapamycin (mTOR) pathway to promote prostate cancer PC3 cell survival; however, the mechanism used by CCL2 to maintain mTOR complex-1 (mTORC1) activation requires clarification. This study demonstrates that upon serum starvation, CCL2 functions as a negative regulator of AMP-activated protein kinase (AMPK) by decreasing phosphorylation at its major regulatory site (Thr(172)) in PC3, DU145, and C4-2B prostate cancer cells. The CCL2-mediated AMPK regulation decreased raptor phosphorylation (Ser(792)) resulting in hyperactivation of mTORC1. D942, a pharmacological activator of AMPK, stunted CCL2-induced mTORC1 activity, survivin expression, and cell survival without significantly affecting Akt activity. CCL2, however, conferred some resistance to the lethal effect of D942 compared with untreated cells. By using Akt-specific inhibitor X, it was shown that Akt inactivation did not cause an increase in AMPK phosphorylation in CCL2-stimulated cells, suggesting that CCL2-mediated negative regulation of AMPK is independent of Akt. Furthermore, bisindolylmaleimide-V, a specific inhibitor of p70(S6K), stunted survivin expression and induced cell death in CCL2-treated PC3. Altogether, these findings suggest that CCL2 hyperactivates mTORC1 through simultaneous regulation of both AMPK and Akt pathways and reveals a new network that promotes prostate cancer: CCL2-AMPK-mTORC1-survivin.
Figures





Similar articles
-
CCL2 protects prostate cancer PC3 cells from autophagic death via phosphatidylinositol 3-kinase/AKT-dependent survivin up-regulation.J Biol Chem. 2008 Sep 5;283(36):25057-73. doi: 10.1074/jbc.M801073200. Epub 2008 Jul 8. J Biol Chem. 2008. PMID: 18611860 Free PMC article.
-
CCL2, survivin and autophagy: new links with implications in human cancer.Autophagy. 2008 Oct;4(7):969-71. doi: 10.4161/auto.6822. Epub 2008 Oct 20. Autophagy. 2008. PMID: 18758234
-
Rapamycin attenuates BAFF-extended proliferation and survival via disruption of mTORC1/2 signaling in normal and neoplastic B-lymphoid cells.J Cell Physiol. 2018 Jan;233(1):516-529. doi: 10.1002/jcp.25913. Epub 2017 May 3. J Cell Physiol. 2018. PMID: 28300280 Free PMC article.
-
LKB1 and AMP-activated protein kinase control of mTOR signalling and growth.Acta Physiol (Oxf). 2009 May;196(1):65-80. doi: 10.1111/j.1748-1716.2009.01972.x. Epub 2009 Feb 19. Acta Physiol (Oxf). 2009. PMID: 19245654 Free PMC article. Review.
-
AMPK and mTOR in cellular energy homeostasis and drug targets.Annu Rev Pharmacol Toxicol. 2012;52:381-400. doi: 10.1146/annurev-pharmtox-010611-134537. Epub 2011 Oct 17. Annu Rev Pharmacol Toxicol. 2012. PMID: 22017684 Review.
Cited by
-
The CCL2 chemokine is a negative regulator of autophagy and necrosis in luminal B breast cancer cells.Breast Cancer Res Treat. 2015 Apr;150(2):309-20. doi: 10.1007/s10549-015-3324-4. Epub 2015 Mar 6. Breast Cancer Res Treat. 2015. PMID: 25744294 Free PMC article.
-
Analysis of tanshinone IIA induced cellular apoptosis in leukemia cells by genome-wide expression profiling.BMC Complement Altern Med. 2012 Jan 16;12:5. doi: 10.1186/1472-6882-12-5. BMC Complement Altern Med. 2012. PMID: 22248096 Free PMC article.
-
CCL2/CCR2 chemokine signaling coordinates survival and motility of breast cancer cells through Smad3 protein- and p42/44 mitogen-activated protein kinase (MAPK)-dependent mechanisms.J Biol Chem. 2012 Oct 19;287(43):36593-608. doi: 10.1074/jbc.M112.365999. Epub 2012 Aug 27. J Biol Chem. 2012. PMID: 22927430 Free PMC article.
-
Loss of transforming growth factor-beta signaling in mammary fibroblasts enhances CCL2 secretion to promote mammary tumor progression through macrophage-dependent and -independent mechanisms.Neoplasia. 2010 May;12(5):425-33. doi: 10.1593/neo.10200. Neoplasia. 2010. PMID: 20454514 Free PMC article.
-
Requirement for ribosomal protein S6 kinase 1 to mediate glycolysis and apoptosis resistance induced by Pten deficiency.Proc Natl Acad Sci U S A. 2011 Feb 8;108(6):2361-5. doi: 10.1073/pnas.1013629108. Epub 2011 Jan 24. Proc Natl Acad Sci U S A. 2011. PMID: 21262837 Free PMC article.
References
-
- Birnbaum MJ. Activating AMP-activated protein kinase without AMP. Mol Cell. 2005;19:289–290. - PubMed
-
- Hawley SA, Pan DA, Mustard KJ, Ross L, Bain J, Edelman AM, Frenguelli BG, Hardie DG. Calmodulin-dependent protein kinase kinase-beta is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab. 2005;2:9–19. - PubMed
-
- Meijer AJ, Codogno P. AMP-activated protein kinase and autophagy. Autophagy. 2007;3:238–240. - PubMed
-
- Meijer AJ, Dubbelhuis PF. Amino acid signalling and the integration of metabolism. Biochem Biophys Res Commun. 2004;313:397–403. - PubMed
-
- Meley D, Bauvy C, Houben-Weerts JH, Dubbelhuis PF, Helmond MT, Codogno P, Meijer AJ. AMP-activated protein kinase and the regulation of autophagic proteolysis. J Biol Chem. 2006;281:34870–34879. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous