

Calmodulin kinase II is required for angiotensin II-mediated vascular smooth muscle hypertrophy
- PMID: 20023119
- PMCID: PMC2822565
- DOI: 10.1152/ajpheart.01014.2009
Calmodulin kinase II is required for angiotensin II-mediated vascular smooth muscle hypertrophy
Abstract
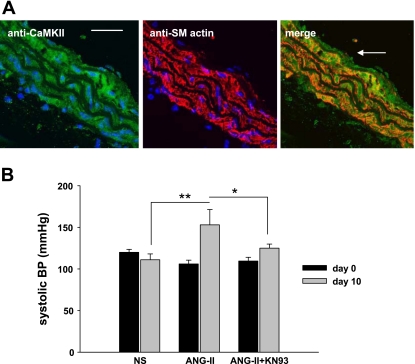
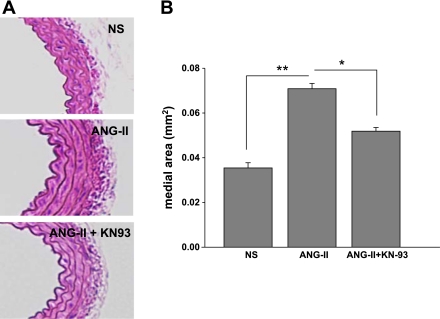
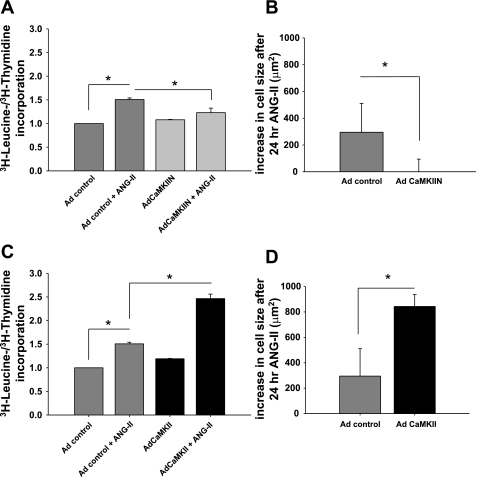
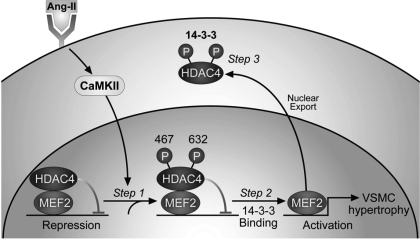
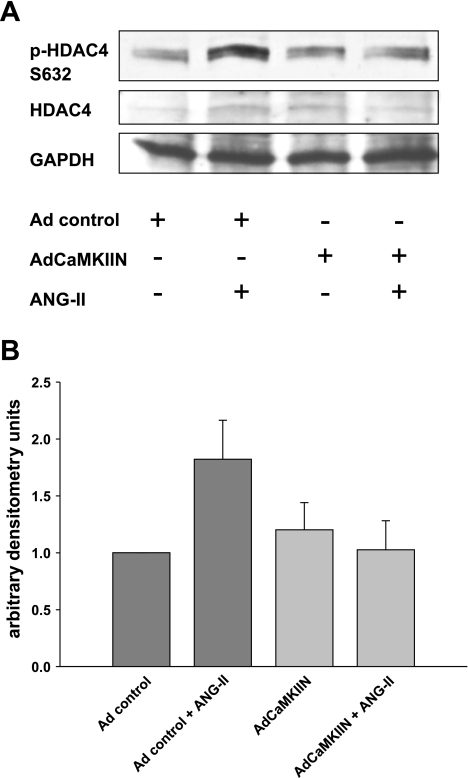
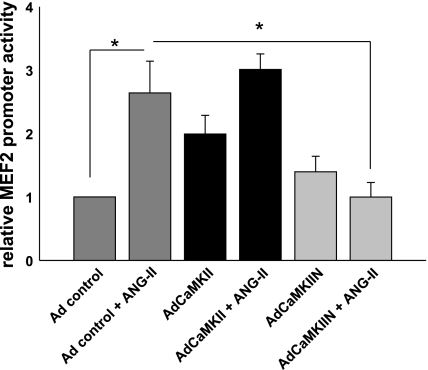
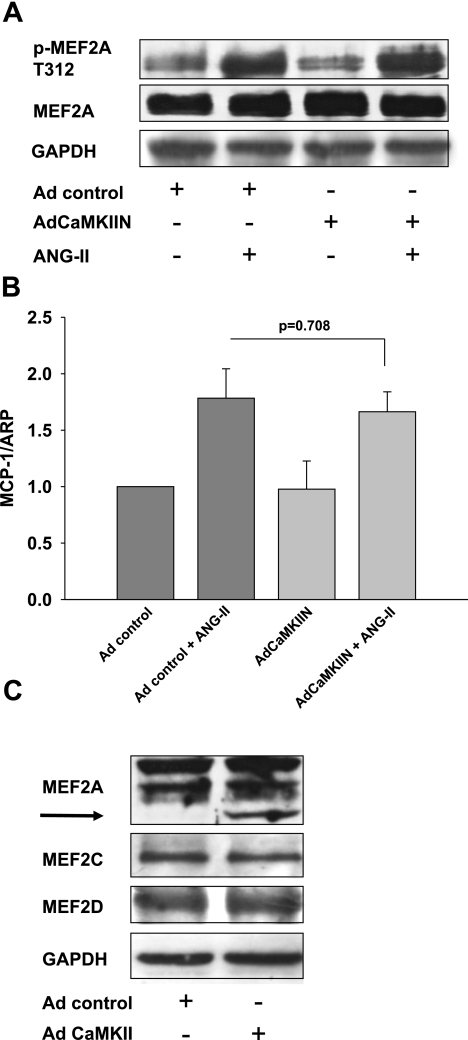
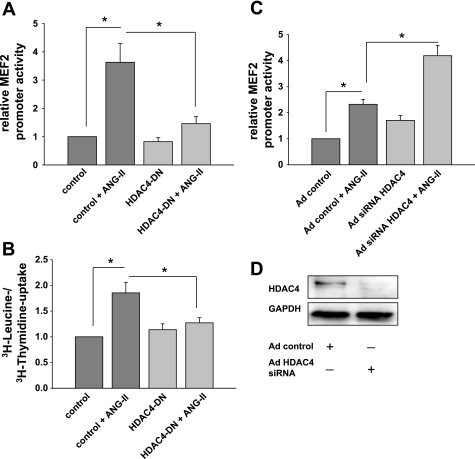
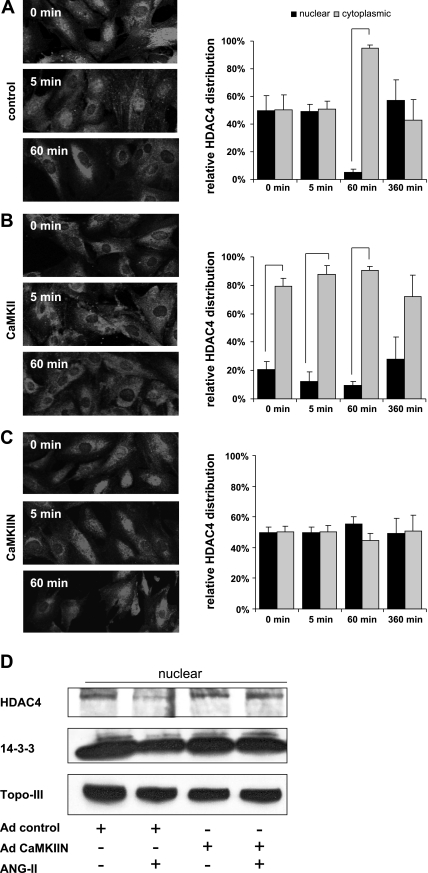
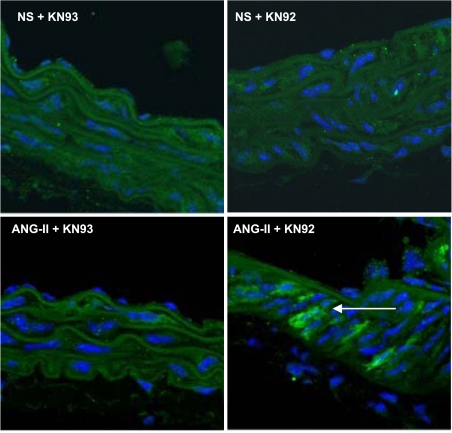
Despite our understanding that medial smooth muscle hypertrophy is a central feature of vascular remodeling, the molecular pathways underlying this pathology are still not well understood. Work over the past decade has illustrated a potential role for the multifunctional calmodulin-dependent kinase CaMKII in smooth muscle cell contraction, growth, and migration. Here we demonstrate that CaMKII is enriched in vascular smooth muscle (VSM) and that CaMKII inhibition blocks ANG II-dependent VSM cell hypertrophy in vitro and in vivo. Specifically, systemic CaMKII inhibition with KN-93 prevented ANG II-mediated hypertension and medial hypertrophy in vivo. Adenoviral transduction with the CaMKII peptide inhibitor CaMKIIN abrogated ANG II-induced VSM hypertrophy in vitro, which was augmented by overexpression of CaMKII-delta2. Finally, we identify the downstream signaling components critical for ANG II- and CaMKII-mediated VSM hypertrophy. Specifically, we demonstrate that CaMKII induces VSM hypertrophy by regulating histone deacetylase 4 (HDAC4) activity, thereby stimulating activity of the hypertrophic transcription factor MEF2. MEF2 transcription is activated by ANG II in vivo and abrogated by the CaMKII inhibitor KN-93. Together, our studies identify a complete pathway for ANG II-triggered arterial VSM hypertrophy and identify new potential therapeutic targets for chronic human hypertension.
Figures
References
-
- Abraham ST, Benscoter H, Schworer CM, Singer HA. In situ Ca2+ dependence for activation of Ca2+/calmodulin-dependent protein kinase II in vascular smooth muscle cells. J Biol Chem 271: 2506–2513, 1996 - PubMed
-
- Abraham ST, Benscoter HA, Schworer CM, Singer HA. A role for Ca2+/calmodulin-dependent protein kinase II in the mitogen-activated protein kinase signaling cascade of cultured rat aortic vascular smooth muscle cells. Circ Res 81: 575–584, 1997 - PubMed
-
- Anderson RD, Haskell RE, Xia H, Roessler BJ, Davidson BL. A simple method for the rapid generation of recombinant adenovirus vectors. Gene Ther 7: 1034–1038, 2000 - PubMed
-
- Backs J, Olson EN. Control of cardiac growth by histone acetylation/deacetylation. Circ Res 98: 15–24, 2006 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous