

Diagnosis of myelodysplastic syndrome among a cohort of 119 patients with fanconi anemia: morphologic and cytogenetic characteristics
- PMID: 20023263
- PMCID: PMC5467447
- DOI: 10.1309/AJCP7W9VMJENZOVG
Diagnosis of myelodysplastic syndrome among a cohort of 119 patients with fanconi anemia: morphologic and cytogenetic characteristics
Abstract
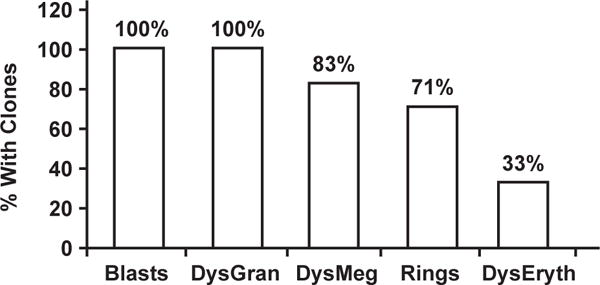
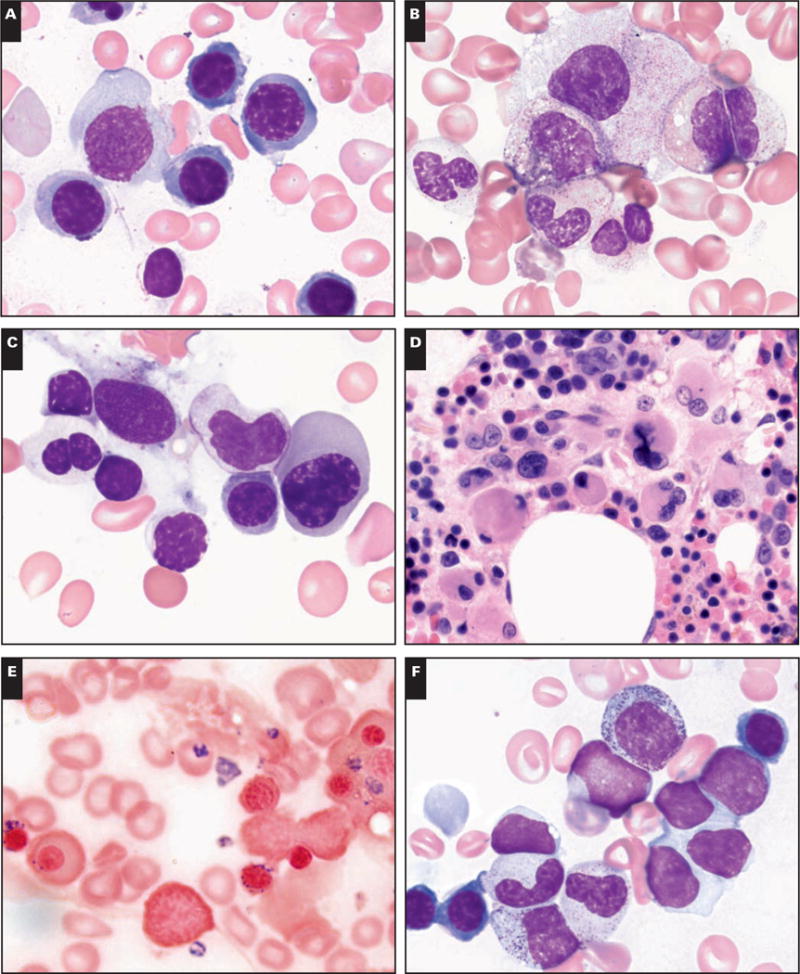
Predisposition to myelodysplastic syndrome (MDS) and acute leukemia is a hallmark of Fanconi anemia (FA). Morphologic criteria for MDS in FA are not well established, nor is the significance of clonal chromosomal abnormalities. We reviewed bone marrow samples of 119 FA patients: 23 had MDS, with the most common subtype refractory cytopenia with multilineage dysplasia. The presence of MDS was highly correlated with the presence of clonal abnormalities. Neutrophil dysplasia and increased blasts were always associated with the presence of a clone, in contrast with dyserythropoiesis. The most frequent clones had gains of 1q and 3q and/or loss of 7. Karyotype complexity also correlated with MDS. One third of patients with 3q as a sole abnormality had no MDS; patients with 3q and an additional abnormality all had MDS. The data provide a rationale for integrating cytogenetic findings with independently evaluated morphologic findings for monitoring bone marrow status in FA.
Figures

Similar articles
-
Numerical chromosomal changes and risk of development of myelodysplastic syndrome--acute myeloid leukemia in patients with Fanconi anemia.Cancer Genet Cytogenet. 2010 Dec;203(2):180-6. doi: 10.1016/j.cancergencyto.2010.07.127. Cancer Genet Cytogenet. 2010. PMID: 21156231
-
Clonal chromosomal aberrations in bone marrow cells of Fanconi anemia patients: gains of the chromosomal segment 3q26q29 as an adverse risk factor.Blood. 2003 May 15;101(10):3872-4. doi: 10.1182/blood-2002-10-3243. Epub 2003 Jan 2. Blood. 2003. PMID: 12511406
-
Chromosome abnormalities in bone marrow of Fanconi anemia patients.Cancer Genet Cytogenet. 1993 Jan;65(1):47-50. doi: 10.1016/0165-4608(93)90057-s. Cancer Genet Cytogenet. 1993. PMID: 8431915
-
[Myelodysplastic syndromes (MDS). Aspects of hematopathologic diagnosis].Pathologe. 2000 Jan;21(1):1-15. doi: 10.1007/s002920050001. Pathologe. 2000. PMID: 10663664 Review. German.
-
Selective pressure as an essential force in molecular evolution of myeloid leukemic clones: a view from the window of Fanconi anemia.Leukemia. 1999 Nov;13(11):1784-9. doi: 10.1038/sj.leu.2401586. Leukemia. 1999. PMID: 10557053 Review.
Cited by
-
Loss of Faap20 Causes Hematopoietic Stem and Progenitor Cell Depletion in Mice Under Genotoxic Stress.Stem Cells. 2015 Jul;33(7):2320-30. doi: 10.1002/stem.2048. Epub 2015 May 25. Stem Cells. 2015. PMID: 25917546 Free PMC article.
-
High Burden of Non-Clonal Chromosome Aberrations Before Onset of Detectable Neoplasia in Fanconi Anemia Bone Marrow.Cancers (Basel). 2025 May 28;17(11):1805. doi: 10.3390/cancers17111805. Cancers (Basel). 2025. PMID: 40507287 Free PMC article.
-
Stat3 and CCAAT enhancer-binding protein β (C/ebpβ) activate Fanconi C gene transcription during emergency granulopoiesis.J Biol Chem. 2018 Mar 16;293(11):3937-3948. doi: 10.1074/jbc.RA117.000528. Epub 2018 Jan 30. J Biol Chem. 2018. PMID: 29382715 Free PMC article.
-
Alternative donor hematopoietic cell transplantation for Fanconi anemia.Blood. 2015 Jun 11;125(24):3798-804. doi: 10.1182/blood-2015-02-626002. Epub 2015 Mar 30. Blood. 2015. PMID: 25824692 Free PMC article. Clinical Trial.
-
Myelodysplastic syndrome: an inability to appropriately respond to damaged DNA?Exp Hematol. 2013 Aug;41(8):665-74. doi: 10.1016/j.exphem.2013.04.008. Epub 2013 Apr 30. Exp Hematol. 2013. PMID: 23643835 Free PMC article. Review.
References
-
- Ameziane N, Errami A, Leveille F, et al. Genetic subtyping of Fanconi anemia by comprehensive mutation screening. Hum Mutat. 2008;29:159–166. - PubMed
-
- Bagby GC, Alter BP. Fanconi anemia. Semin Hematol. 2006;43:147–156. - PubMed
-
- Alter BP. Diagnosis, genetics, and management of inherited bone marrow failure syndromes. Hematology Am Soc Hematol Educ Program. 2007:29–39. - PubMed
-
- Bagby GC, Lipton JM, Sloand EM, et al. Marrow failure. Hematology Am Soc Hematol Educ Program. 2004:318–336. - PubMed
-
- Auerbach AD, Allen RG. Leukemia and preleukemia in Fanconi anemia patients: a review of the literature and report of the International Fanconi Anemia Registry. Cancer Genet Cytogenet. 1991;51:1–12. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
