

A quantitative proteomics analysis of subcellular proteome localization and changes induced by DNA damage
- PMID: 20026476
- PMCID: PMC2849709
- DOI: 10.1074/mcp.M900429-MCP200
A quantitative proteomics analysis of subcellular proteome localization and changes induced by DNA damage
Abstract
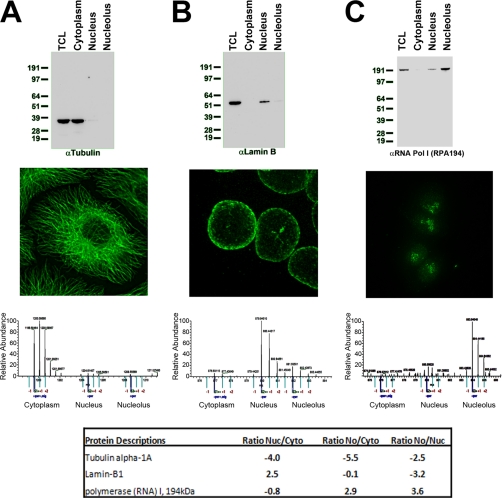
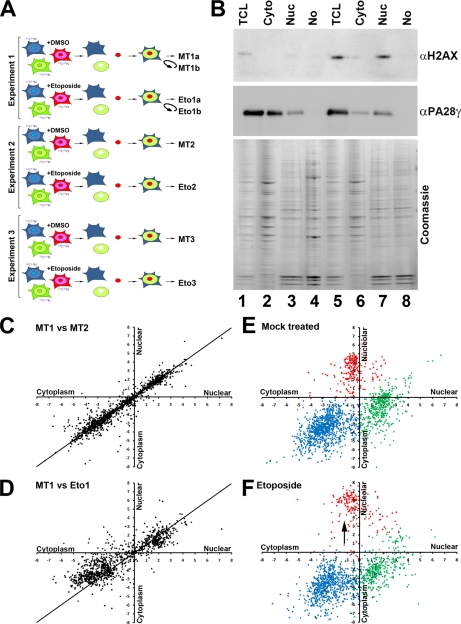
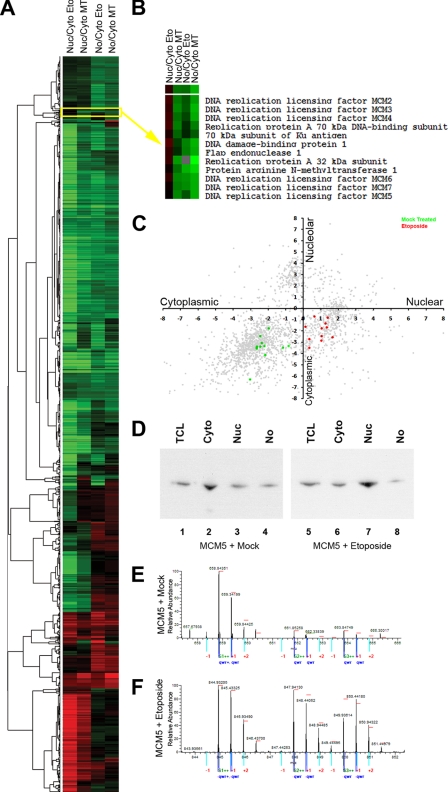
A major challenge in cell biology is to identify the subcellular distribution of proteins within cells and to characterize how protein localization changes under different cell growth conditions and in response to stress and other external signals. Protein localization is usually determined either by microscopy or by using cell fractionation combined with protein blotting techniques. Both these approaches are intrinsically low throughput and limited to the analysis of known components. Here we use mass spectrometry-based proteomics to provide an unbiased, quantitative, and high throughput approach for measuring the subcellular distribution of the proteome, termed "spatial proteomics." The spatial proteomics method analyzes a whole cell extract created by recombining differentially labeled subcellular fractions derived from cells in which proteins have been mass-labeled with heavy isotopes. This was used here to measure the relative distribution between cytoplasm, nucleus, and nucleolus of over 2,000 proteins in HCT116 cells. The data show that, at steady state, the proteome is predominantly partitioned into specific subcellular locations with only a minor subset of proteins equally distributed between two or more compartments. Spatial proteomics also facilitates a proteome-wide comparison of changes in protein localization in response to a wide range of physiological and experimental perturbations, shown here by characterizing dynamic changes in protein localization elicited during the cellular response to DNA damage following treatment of HCT116 cells with etoposide. DNA damage was found to cause dissociation of the proteasome from inhibitory proteins and assembly chaperones in the cytoplasm and relocation to associate with proteasome activators in the nucleus.
Figures



References
-
- Gilchrist A., Au C. E., Hiding J., Bell A. W., Fernandez-Rodriguez J., Lesimple S., Nagaya H., Roy L., Gosline S. J., Hallett M., Paiement J., Kearney R. E., Nilsson T., Bergeron J. J. (2006) Quantitative proteomics analysis of the secretory pathway. Cell 127, 1265–1281 - PubMed
-
- Kislinger T., Cox B., Kannan A., Chung C., Hu P., Ignatchenko A., Scott M. S., Gramolini A. O., Morris Q., Hallett M. T., Rossant J., Hughes T. R., Frey B., Emili A. (2006) Global survey of organ and organelle protein expression in mouse: combined proteomic and transcriptomic profiling. Cell 125, 173–186 - PubMed
-
- Wiese S., Gronemeyer T., Ofman R., Kunze M., Grou C. P., Almeida J. A., Eisenacher M., Stephan C., Hayen H., Schollenberger L., Korosec T., Waterham H. R., Schliebs W., Erdmann R., Berger J., Meyer H. E., Just W., Azevedo J. E., Wanders R. J., Warscheid B. (2007) Proteomics characterization of mouse kidney peroxisomes by tandem mass spectrometry and protein correlation profiling. Mol. Cell. Proteomics 6, 2045–2057 - PubMed
-
- Andersen J. S., Lyon C. E., Fox A. H., Leung A. K., Lam Y. W., Steen H., Mann M., Lamond A. I. (2002) Directed proteomic analysis of the human nucleolus. Curr. Biol 12, 1–11 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
