

Copy number variations in East-Asian population and their evolutionary and functional implications
- PMID: 20026555
- PMCID: PMC2830825
- DOI: 10.1093/hmg/ddp564
Copy number variations in East-Asian population and their evolutionary and functional implications
Abstract
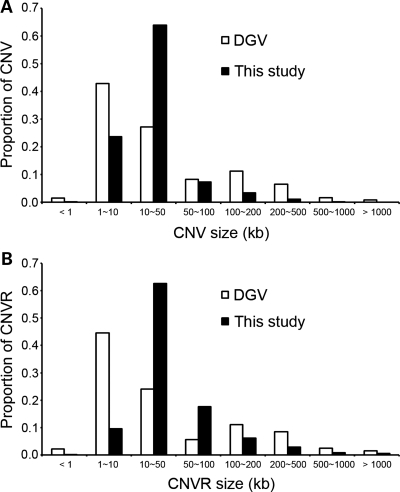
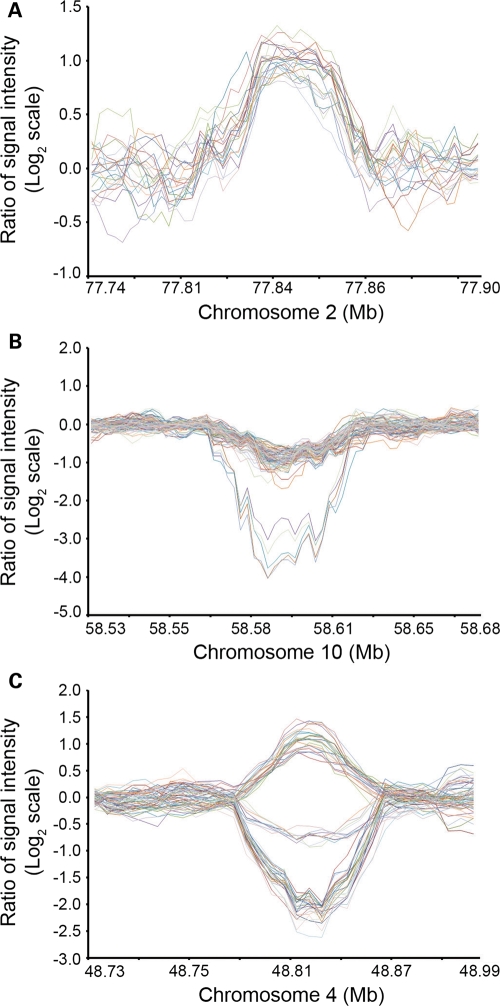
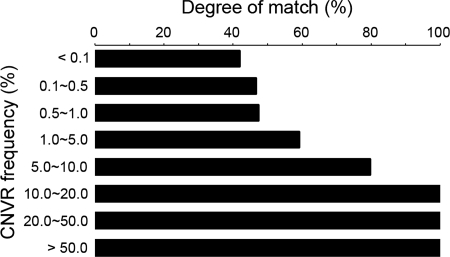
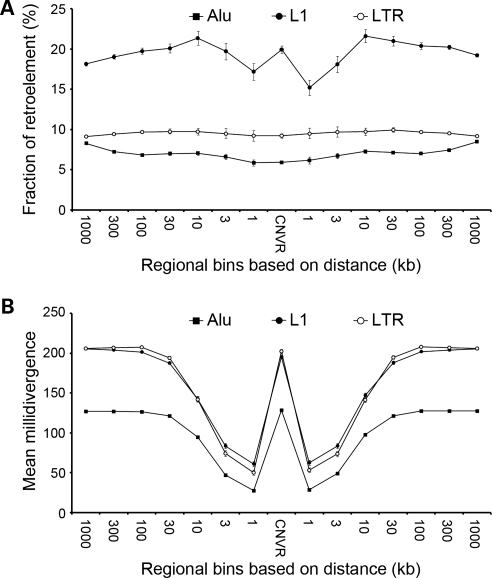
Recent discovery of the copy number variation (CNV) in normal individuals has widened our understanding of genomic variation. However, most of the reported CNVs have been identified in Caucasians, which may not be directly applicable to people of different ethnicities. To profile CNV in East-Asian population, we screened CNVs in 3578 healthy, unrelated Korean individuals, using the Affymetrix Genome-Wide Human SNP array 5.0. We identified 144,207 CNVs using a pooled data set of 100 randomly chosen Korean females as a reference. The average number of CNVs per genome was 40.3, which is higher than that of CNVs previously reported using lower resolution platforms. The median size of CNVs was 18.9 kb (range 0.2-5406 kb). Copy number losses were 4.7 times more frequent than copy number gains. CNV regions (CNVRs) were defined by merging overlapping CNVs identified in two or more samples. In total, 4003 CNVRs were defined encompassing 241.9 Mb accounting for approximately 8% of the human genome. A total of 2077 CNVRs (51.9%) were potentially novel. Known CNVRs were larger and more frequent than novel CNVRs. Sixteen percent of the CNVRs were observed in > or =1% of study subjects and 24% overlapped with the OMIM genes. A total of 476 (11.9%) CNVRs were associated with segmental duplications. CNVS/CNVRs identified in this study will be valuable resources for studying human genome diversity and its association with disease.
Figures
Similar articles
-
The genome-wide landscape of copy number variations in the MUSGEN study provides evidence for a founder effect in the isolated Finnish population.Eur J Hum Genet. 2013 Dec;21(12):1411-6. doi: 10.1038/ejhg.2013.60. Epub 2013 Apr 17. Eur J Hum Genet. 2013. PMID: 23591402 Free PMC article.
-
Inter- and intra-breed genome-wide copy number diversity in a large cohort of European equine breeds.BMC Genomics. 2019 Oct 22;20(1):759. doi: 10.1186/s12864-019-6141-z. BMC Genomics. 2019. PMID: 31640551 Free PMC article.
-
Copy number variations (CNVs) identified in Korean individuals.BMC Genomics. 2008 Oct 18;9:492. doi: 10.1186/1471-2164-9-492. BMC Genomics. 2008. PMID: 18928558 Free PMC article.
-
Genome wide copy number variations using Porcine 60K SNP Beadchip in Landlly pigs.Anim Biotechnol. 2023 Nov;34(6):1891-1899. doi: 10.1080/10495398.2022.2056047. Epub 2022 Apr 4. Anim Biotechnol. 2023. PMID: 35369845 Review.
-
Copy number variants in pharmacogenetic genes.Trends Mol Med. 2011 May;17(5):244-51. doi: 10.1016/j.molmed.2011.01.007. Epub 2011 Mar 8. Trends Mol Med. 2011. PMID: 21388883 Free PMC article. Review.
Cited by
-
CNV analysis in the Lithuanian population.BMC Genet. 2016 May 4;17(1):64. doi: 10.1186/s12863-016-0373-6. BMC Genet. 2016. PMID: 27142071 Free PMC article.
-
Ethnic and functional differentiation of copy number polymorphisms in Tunisian and HapMap population unveils insights on genome organizational plasticity.Sci Rep. 2024 Feb 26;14(1):4654. doi: 10.1038/s41598-024-54749-8. Sci Rep. 2024. PMID: 38409353 Free PMC article.
-
An Update Evolving View of Copy Number Variations in Autoimmune Diseases.Front Genet. 2022 Jan 20;12:794348. doi: 10.3389/fgene.2021.794348. eCollection 2021. Front Genet. 2022. PMID: 35126462 Free PMC article. Review.
-
The genome-wide landscape of copy number variations in the MUSGEN study provides evidence for a founder effect in the isolated Finnish population.Eur J Hum Genet. 2013 Dec;21(12):1411-6. doi: 10.1038/ejhg.2013.60. Epub 2013 Apr 17. Eur J Hum Genet. 2013. PMID: 23591402 Free PMC article.
-
A novel 3-hydroxypropionic acid-inducible promoter regulated by the LysR-type transcriptional activator protein MmsR of Pseudomonas denitrificans.Sci Rep. 2019 Mar 29;9(1):5333. doi: 10.1038/s41598-019-41785-y. Sci Rep. 2019. PMID: 30926872 Free PMC article.
References
-
- Feuk L., Carson A.R., Scherer S.W. Structural variation in the human genome. Nat. Rev. Genet. 2006;7:85–97. - PubMed
-
- Iafrate A.J., Feuk L., Rivera M.N., Listewnik M.L., Donahoe P.K., Qi Y., Scherer S.W., Lee C. Detection of large-scale variation in the human genome. Nat. Genet. 2004;36:949–951. - PubMed
-
- Sebat J., Lakshmi B., Troge J., Alexander J., Young J., Lundin P., Månér S., Massa H., Walker M., Chi M., et al. Large-scale copy number polymorphism in the human genome. Science. 2004;305:525–528. - PubMed
-
- Simon-Sanchez J., Scholz S., Fung H.C., Matarin M., Hernandez D., Gibbs J.R., Britton A., de Vrieze F.W., Peckham E., Gwinn-Hardy K., et al. Genome-wide SNP assay reveals structural genomic variation, extended homozygosity and cell-line induced alterations in normal individuals. Hum. Mol. Genet. 2007;16:1–14. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
