

Exaggerated inflammation, impaired host defense, and neuropathology in progranulin-deficient mice
- PMID: 20026663
- PMCID: PMC2812536
- DOI: 10.1084/jem.20091568
Exaggerated inflammation, impaired host defense, and neuropathology in progranulin-deficient mice
Abstract
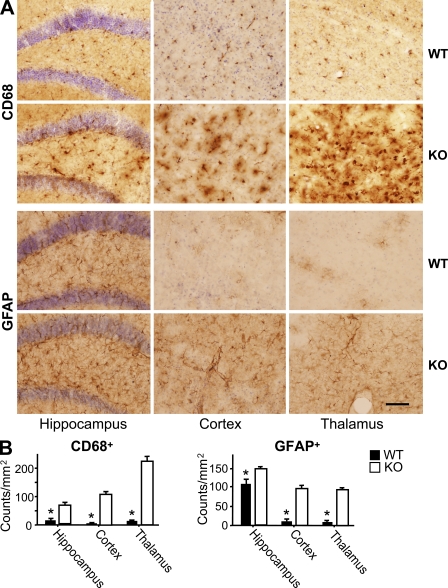
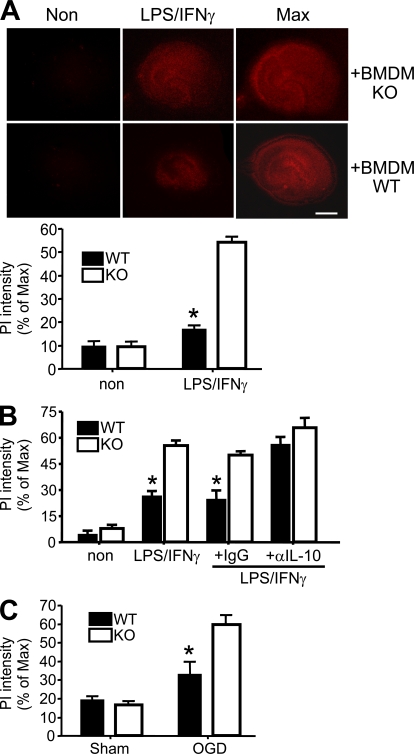
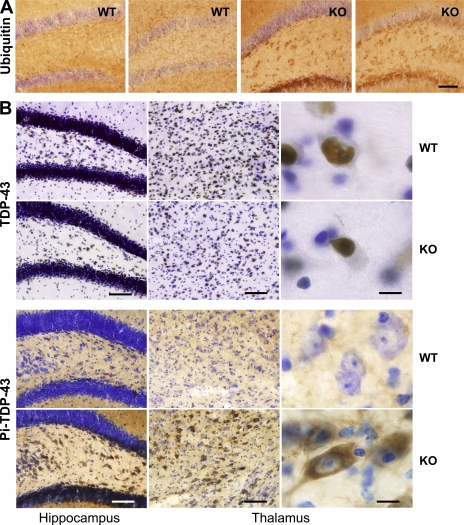
Progranulin (PGRN) is a widely expressed protein involved in diverse biological processes. Haploinsufficiency of PGRN in the human causes tau-negative, ubiquitin-positive frontotemporal dementia (FTD). However, the mechanisms are unknown. To explore the role of PGRN in vivo, we generated PGRN-deficient mice. Macrophages from these mice released less interleukin-10 and more inflammatory cytokines than wild type (WT) when exposed to bacterial lipopolysaccharide. PGRN-deficient mice failed to clear Listeria monocytogenes infection as quickly as WT and allowed bacteria to proliferate in the brain, with correspondingly greater inflammation than in WT. PGRN-deficient macrophages and microglia were cytotoxic to hippocampal cells in vitro, and PGRN-deficient hippocampal slices were hypersusceptible to deprivation of oxygen and glucose. With age, brains of PGRN-deficient mice displayed greater activation of microglia and astrocytes than WT, and their hippocampal and thalamic neurons accumulated cytosolic phosphorylated transactivation response element DNA binding protein-43. Thus, PGRN is a key regulator of inflammation and plays critical roles in both host defense and neuronal integrity. FTD associated with PGRN insufficiency may result from many years of reduced neutrotrophic support together with cumulative damage in association with dysregulated inflammation.
Figures
References
-
- Arai T., Hasegawa M., Akiyama H., Ikeda K., Nonaka T., Mori H., Mann D., Tsuchiya K., Yoshida M., Hashizume Y., Oda T. 2006. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 351:602–611 10.1016/j.bbrc.2006.10.093 - DOI - PubMed
-
- Björkqvist M., Wild E.J., Thiele J., Silvestroni A., Andre R., Lahiri N., Raibon E., Lee R.V., Benn C.L., Soulet D., et al. 2008. A novel pathogenic pathway of immune activation detectable before clinical onset in Huntington’s disease. J. Exp. Med. 205:1869–1877 10.1084/jem.20080178 - DOI - PMC - PubMed
-
- Cairns N.J., Neumann M., Bigio E.H., Holm I.E., Troost D., Hatanpaa K.J., Foong C., White C.L., III, Schneider J.A., Kretzschmar H.A., et al. 2007. TDP-43 in familial and sporadic frontotemporal lobar degeneration with ubiquitin inclusions. Am. J. Pathol. 171:227–240 10.2353/ajpath.2007.070182 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
