

The role of the Akt/mTOR pathway in tobacco carcinogen-induced lung tumorigenesis
- PMID: 20028747
- PMCID: PMC2805044
- DOI: 10.1158/1078-0432.CCR-09-0234
The role of the Akt/mTOR pathway in tobacco carcinogen-induced lung tumorigenesis
Abstract
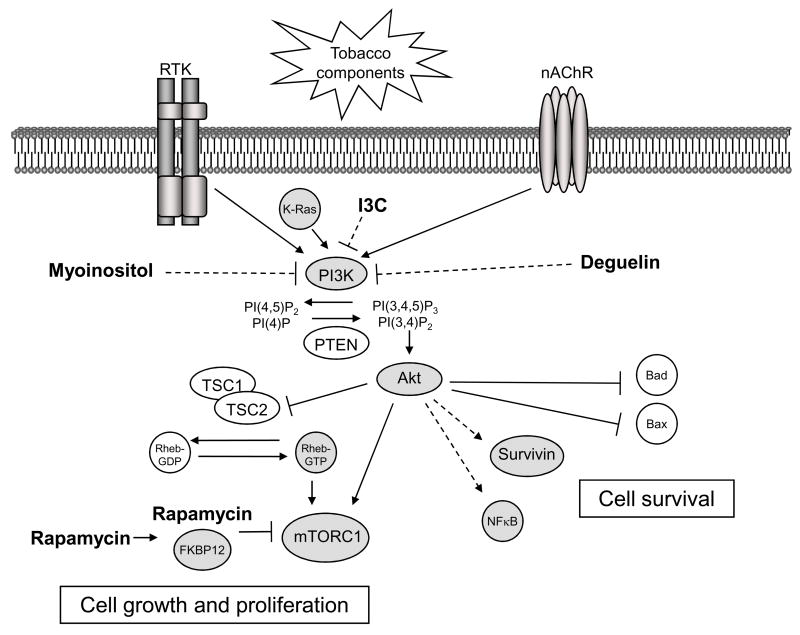
Lung cancer is the leading cause of cancer-related death in the United States, and 85 to 90% of lung cancer cases are associated with tobacco use. Tobacco components promote lung tumorigenesis through genotoxic effects, as well as through biochemical modulation of signaling pathways such as the Akt/mammalian target of rapamycin (mTOR) pathway that regulates cell proliferation and survival. This review will describe cell surface receptors and other upstream components required for tobacco carcinogen-induced activation of Akt and mTOR. Preclinical studies show that inhibitors of the Akt/mTOR pathway inhibit tumor formation in mouse models of carcinogen-induced lung tumorigenesis. Some of these inhibitors will be highlighted, and their clinical potential for the treatment and prevention of lung cancer will be discussed.
Figures
Similar articles
-
New inhibitors of the mammalian target of rapamycin signaling pathway for cancer.Expert Opin Investig Drugs. 2010 Aug;19(8):919-30. doi: 10.1517/13543784.2010.499121. Expert Opin Investig Drugs. 2010. PMID: 20569080 Review.
-
Dual inhibition of akt/mammalian target of rapamycin pathway by nanoparticle albumin-bound-rapamycin and perifosine induces antitumor activity in multiple myeloma.Mol Cancer Ther. 2010 Apr;9(4):963-75. doi: 10.1158/1535-7163.MCT-09-0763. Epub 2010 Apr 6. Mol Cancer Ther. 2010. PMID: 20371718 Free PMC article.
-
Fibronectin stimulates non-small cell lung carcinoma cell growth through activation of Akt/mammalian target of rapamycin/S6 kinase and inactivation of LKB1/AMP-activated protein kinase signal pathways.Cancer Res. 2006 Jan 1;66(1):315-23. doi: 10.1158/0008-5472.CAN-05-2367. Cancer Res. 2006. PMID: 16397245
-
[Targeting of the AKT-mTOR pathway in head and neck and lung cancer].Bull Cancer. 2009;96 Suppl 1:S57-63. doi: 10.1684/bdc.2009.0787. Bull Cancer. 2009. PMID: 19433374 Review. French.
-
CGP57380 enhances efficacy of RAD001 in non-small cell lung cancer through abrogating mTOR inhibition-induced phosphorylation of eIF4E and activating mitochondrial apoptotic pathway.Oncotarget. 2016 May 10;7(19):27787-801. doi: 10.18632/oncotarget.8497. Oncotarget. 2016. PMID: 27050281 Free PMC article.
Cited by
-
Bioactive PI3-kinase/Akt/mTOR Inhibitors in Targeted Lung Cancer Therapy.Adv Pharm Bull. 2023 Jan;13(1):24-35. doi: 10.34172/apb.2023.003. Epub 2021 Oct 9. Adv Pharm Bull. 2023. PMID: 36721812 Free PMC article. Review.
-
Chemoprevention of lung cancer: prospects and disappointments in human clinical trials.Cancers (Basel). 2013 Jan 24;5(1):131-48. doi: 10.3390/cancers5010131. Cancers (Basel). 2013. PMID: 24216701 Free PMC article.
-
The Akt1/IL-6/STAT3 pathway regulates growth of lung tumor initiating cells.Oncotarget. 2015 Dec 15;6(40):42667-86. doi: 10.18632/oncotarget.5626. Oncotarget. 2015. PMID: 26486080 Free PMC article.
-
Dihydrocapsaicin Inhibits Epithelial Cell Transformation through Targeting Amino Acid Signaling and c-Fos Expression.Nutrients. 2019 Jun 4;11(6):1269. doi: 10.3390/nu11061269. Nutrients. 2019. PMID: 31167465 Free PMC article.
-
Insights into the Mechanisms of Action of Proanthocyanidins and Anthocyanins in the Treatment of Nicotine-Induced Non-Small Cell Lung Cancer.Int J Mol Sci. 2022 Jul 18;23(14):7905. doi: 10.3390/ijms23147905. Int J Mol Sci. 2022. PMID: 35887251 Free PMC article. Review.
References
-
- Catassi A, Servent D, Paleari L, Cesario A, Russo P. Multiple roles of nicotine on cell proliferation and inhibition of apoptosis: implications on lung carcinogenesis. Mutat Res. 2008;659:221–31. - PubMed
-
- Clark AS, West KA, Blumberg PM, Dennis PA. Altered protein kinase C (PKC) isoforms in non-small cell lung cancer cells: PKCdelta promotes cellular survival and chemotherapeutic resistance. Cancer Res. 2003;63:780–6. - PubMed
-
- Jull BA, Plummer HK, 3rd, Schuller HM. Nicotinic receptor-mediated activation by the tobacco-specific nitrosamine NNK of a Raf-1/MAP kinase pathway, resulting in phosphorylation of c-myc in human small cell lung carcinoma cells and pulmonary neuroendocrine cells. J Cancer Res Clin Oncol. 2001;127:707–17. - PubMed
-
- Arredondo J, Chernyavsky AI, Jolkovsky DL, Pinkerton KE, Grando SA. Receptor-mediated tobacco toxicity: cooperation of the Ras/Raf-1/MEK1/ERK and JAK-2/STAT-3 pathways downstream of alpha7 nicotinic receptor in oral keratinocytes. FASEB J. 2006;20:2093–101. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
