

Clinical and functional characterization of TNNT2 mutations identified in patients with dilated cardiomyopathy
- PMID: 20031601
- PMCID: PMC2900844
- DOI: 10.1161/CIRCGENETICS.108.846733
Clinical and functional characterization of TNNT2 mutations identified in patients with dilated cardiomyopathy
Abstract
Background: A key issue for cardiovascular genetic medicine is ascertaining if a putative mutation indeed causes dilated cardiomyopathy (DCM). This is critically important as genetic DCM, usually presenting with advanced, life-threatening disease, may be preventable with early intervention in relatives known to carry the mutation.
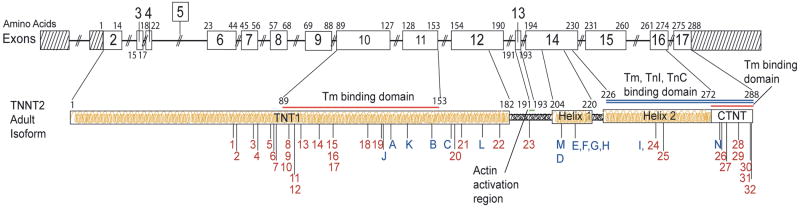
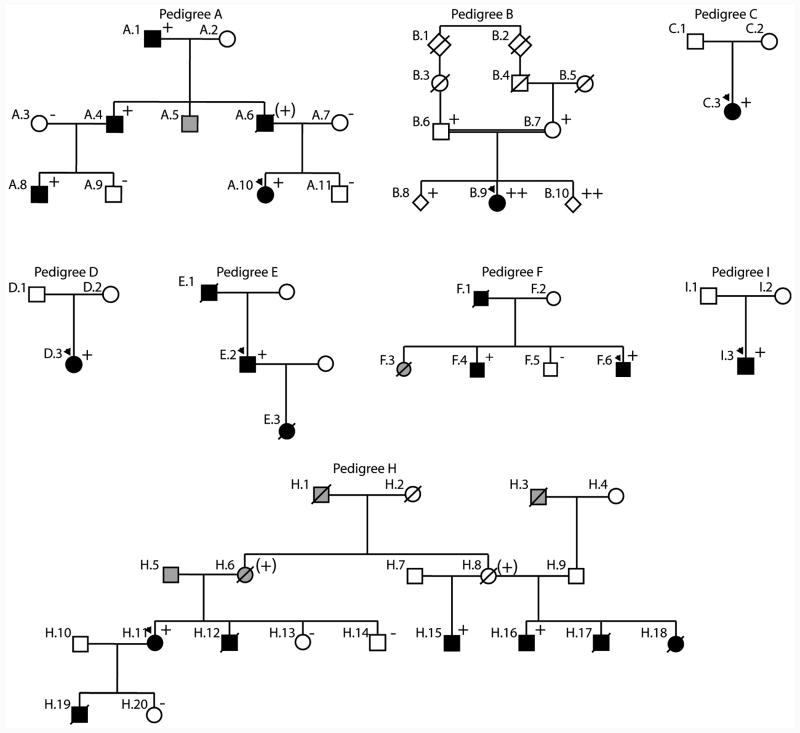
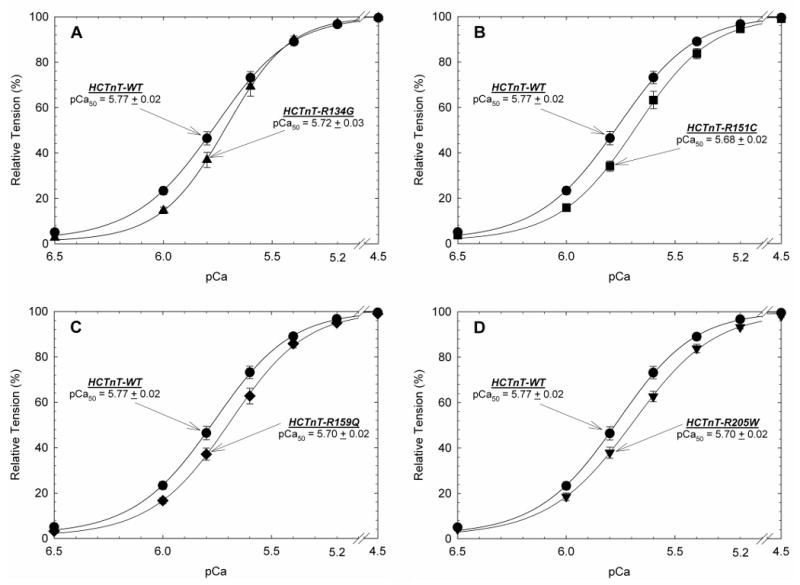
Methods and results: We recently undertook bidirectional resequencing of TNNT2, the cardiac troponin T gene, in 313 probands with DCM. We identified 6 TNNT2 protein-altering variants in 9 probands, all who had early onset, aggressive disease. Additional family members of mutation carriers were then studied when available. Four of the 9 probands had DCM without a family history, and 5 probands had familial DCM. Only 1 mutation (Lys210del) could be attributed as definitively causative from previous reports. Four of the 5 missense mutations were novel (Arg134Gly, Arg151Cys, Arg159Gln, and Arg205Trp), and one was previously reported with hypertrophic cardiomyopathy (Glu244Asp). Based on the clinical, pedigree, and molecular genetic data, these 5 mutations were considered possibly or likely disease causing. To further clarify their potential pathophysiologic impact, we undertook functional studies of these mutations in cardiac myocytes reconstituted with mutant troponin T proteins. We observed decreased Ca(2+) sensitivity of force development, a hallmark of DCM, in support of the conclusion that these mutations are disease causing.
Conclusions: We conclude that the combination of clinical, pedigree, molecular genetic, and functional data strengthen the interpretation of TNNT2 mutations in DCM.
Conflict of interest statement
Figures

References
-
- Hershberger R, Lindenfeld J, Mestroni L, Seidman C, Taylor M, Towbin J. Genetic Evaluation of Cardiomyopathy - A Heart Failure Society of America Practice Guideline. J Cardiac Failure. 2009;15:83–97. - PubMed
-
- Burkett EL, Hershberger RE. Clinical and genetic issues in familial dilated cardiomyopathy. J Am Coll Cardiol. 2005;45:969–81. - PubMed
-
- Ashrafian H, Watkins H. Reviews of translational medicine and genomics in cardiovascular disease: new disease taxonomy and therapeutic implications cardiomyopathies: therapeutics based on molecular phenotype. J Am Coll Cardiol. 2007;49:1251–64. - PubMed
-
- Fatkin D, MacRae C, Sasaki T, Wolff M, Porcu M, Frenneaux M, Atherton J, Vidaillet H, Spudich S, Girolami U, Seidman J, Seidman C. Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction-system disease. N Engl J Med. 1999;341:1715–24. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
