

Inhibition of calcineurin-mediated endocytosis and alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors prevents amyloid beta oligomer-induced synaptic disruption
- PMID: 20032460
- PMCID: PMC2844209
- DOI: 10.1074/jbc.M109.057182
Inhibition of calcineurin-mediated endocytosis and alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors prevents amyloid beta oligomer-induced synaptic disruption
Abstract
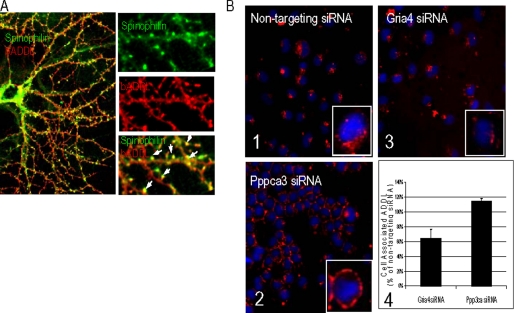
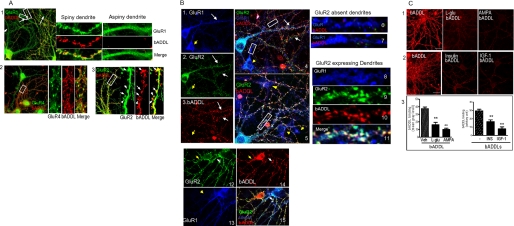
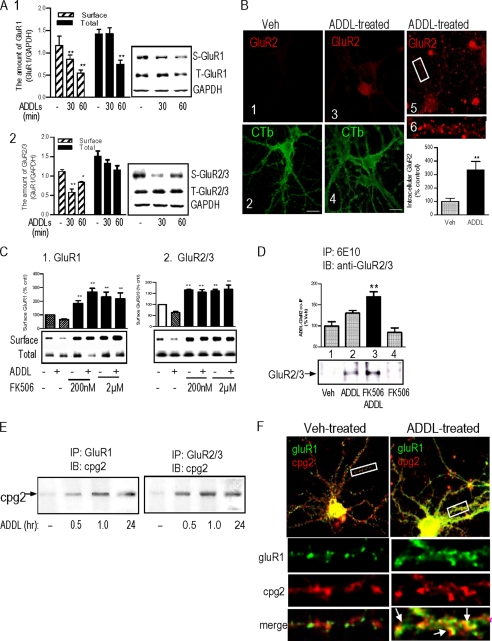
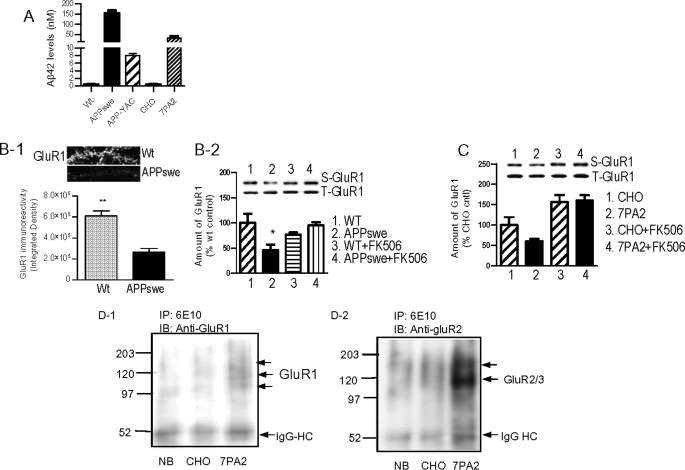
Synaptic degeneration, including impairment of synaptic plasticity and loss of synapses, is an important feature of Alzheimer disease pathogenesis. Increasing evidence suggests that these degenerative synaptic changes are associated with an accumulation of soluble oligomeric assemblies of amyloid beta (Abeta) known as ADDLs. In primary hippocampal cultures ADDLs bind to a subpopulation of neurons. However the molecular basis of this cell type-selective interaction is not understood. Here, using siRNA screening technology, we identified alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor subunits and calcineurin as candidate genes potentially involved in ADDL-neuron interactions. Immunocolocalization experiments confirmed that ADDL binding occurs in dendritic spines that express surface AMPA receptors, particularly the calcium-impermeable type II AMPA receptor subunit (GluR2). Pharmacological removal of the surface AMPA receptors or inhibition of AMPA receptors with antagonists reduces ADDL binding. Furthermore, using co-immunoprecipitation and photoreactive amino acid cross-linking, we found that ADDLs interact preferentially with GluR2-containing complexes. We demonstrate that calcineurin mediates an endocytotic process that is responsible for the rapid internalization of bound ADDLs along with surface AMPA receptor subunits, which then both colocalize with cpg2, a molecule localized specifically at the postsynaptic endocytic zone of excitatory synapses that plays an important role in activity-dependent glutamate receptor endocytosis. Both AMPA receptor and calcineurin inhibitors prevent oligomer-induced surface AMPAR and spine loss. These results support a model of disease pathogenesis in which Abeta oligomers interact selectively with neurotransmission pathways at excitatory synapses, resulting in synaptic loss via facilitated endocytosis. Validation of this model in human disease would identify therapeutic targets for Alzheimer disease.
Figures




References
-
- Selkoe D. J. (2002) Science 298, 789–791 - PubMed
-
- Coleman P. D., Yao P. J. (2003) Neurobiol. Aging 24, 1023–1027 - PubMed
-
- Lassmann H., Fischer P., Jellinger K. (1993) Ann. N.Y. Acad. Sci. 695, 59–64 - PubMed
-
- Terry R. D., Masliah E., Salmon D. P., Butters N., DeTeresa R., Hill R., Hansen L. A., Katzman R. (1991) Ann. Neurol. 30, 572–580 - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
