

Joubert syndrome 2 (JBTS2) in Ashkenazi Jews is associated with a TMEM216 mutation
- PMID: 20036350
- PMCID: PMC2801745
- DOI: 10.1016/j.ajhg.2009.12.007
Joubert syndrome 2 (JBTS2) in Ashkenazi Jews is associated with a TMEM216 mutation
Erratum in
- Am J Hum Genet. 2010 Feb;86(2):294. Shanske, Alan L [added]
Abstract
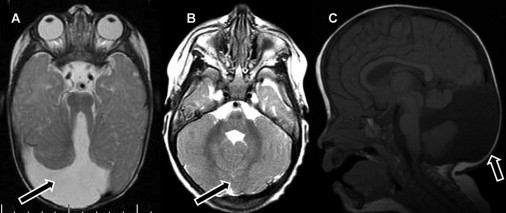
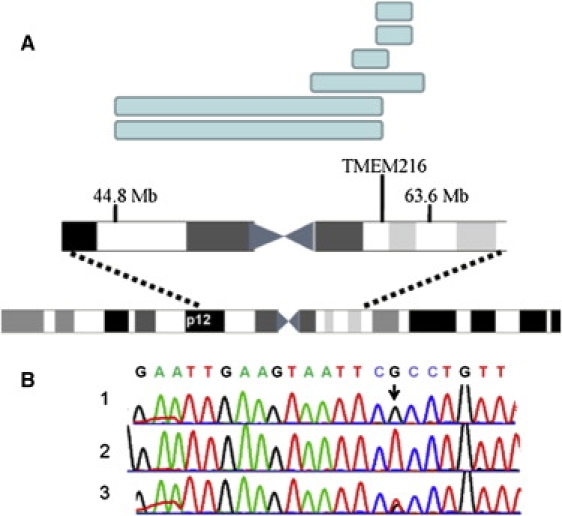
Patients with Joubert syndrome 2 (JBTS2) suffer from a neurological disease manifested by psychomotor retardation, hypotonia, ataxia, nystagmus, and oculomotor apraxia and variably associated with dysmorphism, as well as retinal and renal involvement. Brain MRI results show cerebellar vermis hypoplasia and additional anomalies of the fourth ventricle, corpus callosum, and occipital cortex. The disease has previously been mapped to the centromeric region of chromosome 11. Using homozygosity mapping in 13 patients from eight Ashkenazi Jewish families, we identified a homozygous mutation, R12L, in the TMEM216 gene, in all affected individuals. Thirty individuals heterozygous for the mutation were detected among 2766 anonymous Ashkenazi Jews, indicating a carrier rate of 1:92. Given the small size of the TMEM216 gene relative to other JBTS genes, its sequence analysis is warranted in all JBTS patients, especially those who suffer from associated anomalies.
2010 The American Society of Human Genetics. Published by Elsevier Inc.
Figures
References
-
- Satran D., Pierpont M.E., Dobyns W.B. Cerebello-oculo-renal syndromes including Arima, Senior-Löken and COACH syndromes: more than just variants of Joubert syndrome. Am. J. Med. Genet. 1999;86:459–469. - PubMed
-
- Verloes A., Lambotte C. Further delineation of a syndrome of cerebellar vermis hypo/aplasia, oligophrenia, congenital ataxia, coloboma, and hepatic fibrosis. Am. J. Med. Genet. 1989;32:227–232. - PubMed
-
- Haug K., Khan S., Fuchs S., König R. OFD II, OFD VI, and Joubert syndrome manifestations in 2 sibs. Am. J. Med. Genet. 2000;91:135–137. - PubMed
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
