

The antiviral factor APOBEC3G improves CTL recognition of cultured HIV-infected T cells
- PMID: 20038599
- PMCID: PMC2812543
- DOI: 10.1084/jem.20091933
The antiviral factor APOBEC3G improves CTL recognition of cultured HIV-infected T cells
Abstract
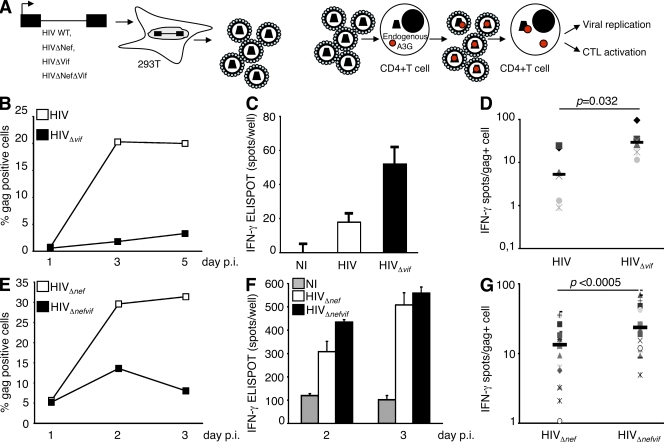
The cytidine deaminase APOBEC3G (A3G) enzyme exerts an intrinsic anti-human immunodeficiency virus (HIV) defense by introducing lethal G-to-A hypermutations in the viral genome. The HIV-1 viral infectivity factor (Vif) protein triggers degradation of A3G and counteracts this antiviral effect. The impact of A3G on the adaptive cellular immune response has not been characterized. We examined whether A3G-edited defective viruses, which are known to express truncated or misfolded viral proteins, activate HIV-1-specific (HS) CD8+ cytotoxic T lymphocytes (CTLs). To this end, we compared the immunogenicity of cells infected with wild-type or Vif-deleted viruses in the presence or absence of the cytidine deaminase. The inhibitory effect of A3G on HIV replication was associated with a strong activation of cocultivated HS-CTLs. CTL activation was particularly marked with Vif-deleted HIV and with viruses harboring A3G. Enzymatically inactive A3G mutants failed to enhance CTL activation. We also engineered proviruses bearing premature stop codons in their genome as scars of A3G editing. These viruses were not infectious but potently activated HS-CTLs. Therefore, the pool of defective viruses generated by A3G represents an underestimated source of viral antigens. Our results reveal a novel function for A3G, acting not only as an intrinsic antiviral factor but also as an inducer of the adaptive immune system.
Figures




Similar articles
-
APOBEC3G and APOBEC3F Act in Concert To Extinguish HIV-1 Replication.J Virol. 2016 Apr 14;90(9):4681-4695. doi: 10.1128/JVI.03275-15. Print 2016 May. J Virol. 2016. PMID: 26912618 Free PMC article.
-
Small-molecule inhibition of HIV-1 Vif.Nat Biotechnol. 2008 Oct;26(10):1187-92. doi: 10.1038/nbt.1496. Epub 2008 Sep 21. Nat Biotechnol. 2008. PMID: 18806783 Free PMC article.
-
Long-term passage of Vif-null HIV-1 in CD4+ T cells expressing sub-lethal levels of APOBEC proteins fails to develop APOBEC resistance.Virology. 2017 Apr;504:1-11. doi: 10.1016/j.virol.2017.01.016. Epub 2017 Jan 25. Virology. 2017. PMID: 28131088 Free PMC article.
-
APOBEC3G: an intracellular centurion.Philos Trans R Soc Lond B Biol Sci. 2009 Mar 12;364(1517):689-703. doi: 10.1098/rstb.2008.0193. Philos Trans R Soc Lond B Biol Sci. 2009. PMID: 19008196 Free PMC article. Review.
-
Various strategies for developing APOBEC3G protectors to circumvent human immunodeficiency virus type 1.Eur J Med Chem. 2023 Mar 15;250:115188. doi: 10.1016/j.ejmech.2023.115188. Epub 2023 Feb 6. Eur J Med Chem. 2023. PMID: 36773550 Review.
Cited by
-
APOBEC3 proteins can copackage and comutate HIV-1 genomes.Nucleic Acids Res. 2016 Sep 19;44(16):7848-65. doi: 10.1093/nar/gkw653. Epub 2016 Jul 20. Nucleic Acids Res. 2016. PMID: 27439715 Free PMC article.
-
Convergent Evolution of HLA-C Downmodulation in HIV-1 and HIV-2.mBio. 2020 Jul 14;11(4):e00782-20. doi: 10.1128/mBio.00782-20. mBio. 2020. PMID: 32665270 Free PMC article.
-
The cellular antiviral protein APOBEC3G interacts with HIV-1 reverse transcriptase and inhibits its function during viral replication.J Virol. 2012 Apr;86(7):3777-86. doi: 10.1128/JVI.06594-11. Epub 2012 Feb 1. J Virol. 2012. PMID: 22301159 Free PMC article.
-
Possible footprints of APOBEC3F and/or other APOBEC3 deaminases, but not APOBEC3G, on HIV-1 from patients with acute/early and chronic infections.J Virol. 2014 Nov;88(21):12882-94. doi: 10.1128/JVI.01460-14. Epub 2014 Aug 27. J Virol. 2014. PMID: 25165112 Free PMC article.
-
Longitudinal study reveals HIV-1-infected CD4+ T cell dynamics during long-term antiretroviral therapy.J Clin Invest. 2020 Jul 1;130(7):3543-3559. doi: 10.1172/JCI135953. J Clin Invest. 2020. PMID: 32191639 Free PMC article. Clinical Trial.
References
-
- Armitage A.E., Katzourakis A., de Oliveira T., Welch J.J., Belshaw R., Bishop K.N., Kramer B., McMichael A.J., Rambaut A., Iversen A.K. 2008. Conserved footprints of APOBEC3G on Hypermutated human immunodeficiency virus type 1 and human endogenous retrovirus HERV-K(HML2) sequences. J. Virol. 82:8743–8761 10.1128/JVI.00584-08 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
