

Compounds from an unbiased chemical screen reverse both ER-to-Golgi trafficking defects and mitochondrial dysfunction in Parkinson's disease models
- PMID: 20038714
- PMCID: PMC2869493
- DOI: 10.1242/dmm.004267
Compounds from an unbiased chemical screen reverse both ER-to-Golgi trafficking defects and mitochondrial dysfunction in Parkinson's disease models
Abstract
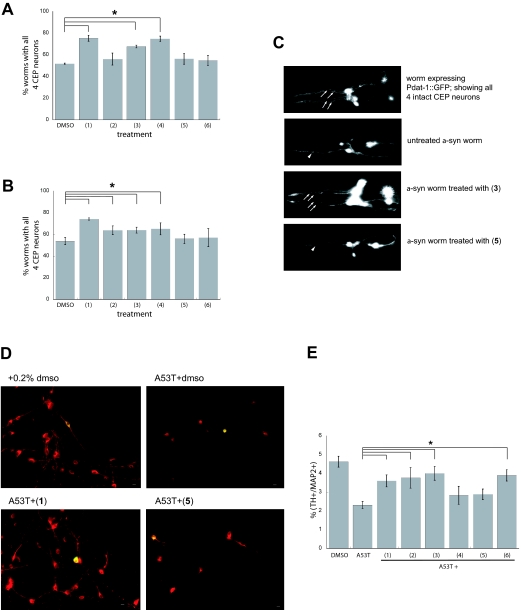
alpha-Synuclein (alpha-syn) is a small lipid-binding protein involved in vesicle trafficking whose function is poorly characterized. It is of great interest to human biology and medicine because alpha-syn dysfunction is associated with several neurodegenerative disorders, including Parkinson's disease (PD). We previously created a yeast model of alpha-syn pathobiology, which established vesicle trafficking as a process that is particularly sensitive to alpha-syn expression. We also uncovered a core group of proteins with diverse activities related to alpha-syn toxicity that is conserved from yeast to mammalian neurons. Here, we report that a yeast strain expressing a somewhat higher level of alpha-syn also exhibits strong defects in mitochondrial function. Unlike our previous strain, genetic suppression of endoplasmic reticulum (ER)-to-Golgi trafficking alone does not suppress alpha-syn toxicity in this strain. In an effort to identify individual compounds that could simultaneously rescue these apparently disparate pathological effects of alpha-syn, we screened a library of 115,000 compounds. We identified a class of small molecules that reduced alpha-syn toxicity at micromolar concentrations in this higher toxicity strain. These compounds reduced the formation of alpha-syn foci, re-established ER-to-Golgi trafficking and ameliorated alpha-syn-mediated damage to mitochondria. They also corrected the toxicity of alpha-syn in nematode neurons and in primary rat neuronal midbrain cultures. Remarkably, the compounds also protected neurons against rotenone-induced toxicity, which has been used to model the mitochondrial defects associated with PD in humans. That single compounds are capable of rescuing the diverse toxicities of alpha-syn in yeast and neurons suggests that they are acting on deeply rooted biological processes that connect these toxicities and have been conserved for a billion years of eukaryotic evolution. Thus, it seems possible to develop novel therapeutic strategies to simultaneously target the multiple pathological features of PD.
Figures





Comment in
-
Preventing Parkinson's pathology.Dis Model Mech. 2010 Jul-Aug;3(7-8):399-400. doi: 10.1242/dmm.005678. Epub 2010 May 18. Dis Model Mech. 2010. PMID: 20483997 No abstract available.
References
-
- Abeliovich A, Schmitz Y, Farinas I, Choi-Lundberg D, Ho WH, Castillo PE, Shinsky N, Verdugo JM, Armanini M, Ryan A, et al. (2000). Mice lacking alpha-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron 25, 239–252 - PubMed
-
- Abou-Sleiman PM, Muqit MM, Wood NW. (2006). Expanding insights of mitochondrial dysfunction in Parkinson’s disease. Nat Rev Neurosci. 7, 207–219 - PubMed
-
- Attfield PV. (1997). Stress tolerance: the key to effective strains of industrial baker’s yeast. Nat Biotechnol 15, 1351–1357 - PubMed
-
- Ayala A, Venero JL, Cano J, Machado A. (2007). Mitochondrial toxins and neurodegenerative diseases. Front Biosci. 12, 986–1007 - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
