

Autophagy in skeletal muscle: implications for Pompe disease
- PMID: 20040311
- PMCID: PMC2948975
- DOI: 10.5414/cpp47042
Autophagy in skeletal muscle: implications for Pompe disease
Abstract
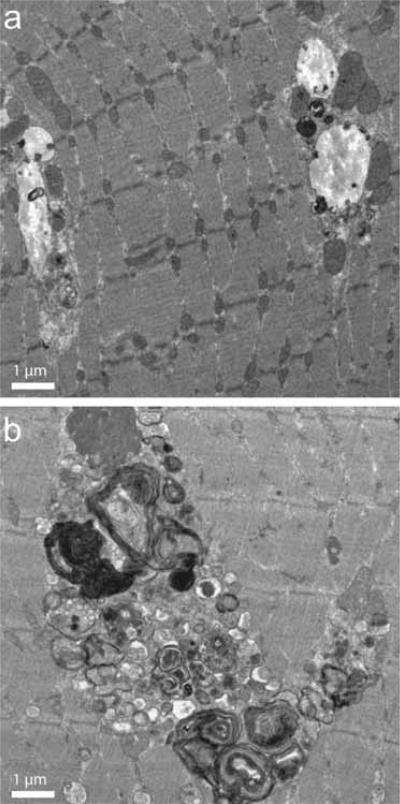
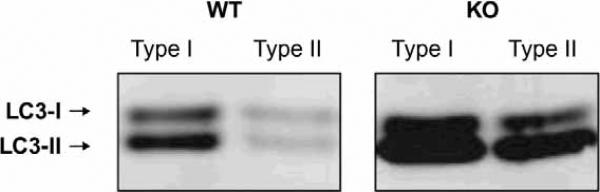
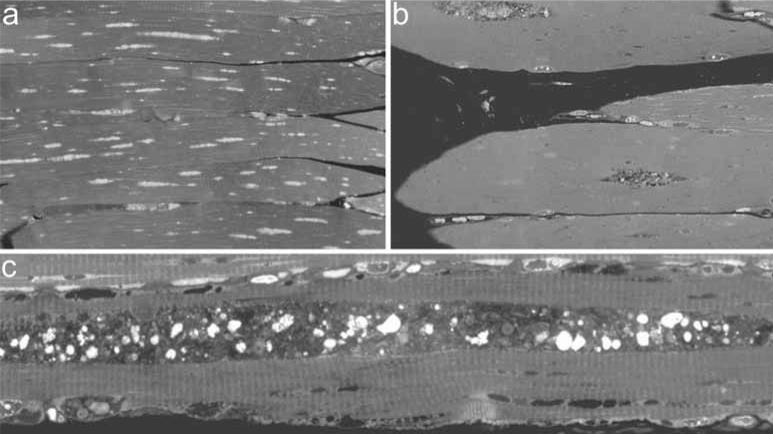
Pompe disease is caused by an inherited deficiency of acid a-glucosidase (GAA), a lysosomal enzyme that catalyzes the breakdown of glycogen to glucose. In the absence of GAA, enlarged, glycogen-laden lysosomes accumulate in multiple tissues, although the major clinical manifestations are seen in cardiac and skeletal muscle. For many years, it was believed that the rupture of glycogen-filled lysosomes was the major cause of the profound muscle damage observed in patients with Pompe disease. Here, we present evidence that a failure of productive autophagy in muscle tissue contributes strongly to disease pathology in both patients with Pompe disease and GAA-knockout mice. In the GAA-knockout mouse model, progressive accumulation of autophagic vesicles is restricted to Type II-rich muscle fibers. Not only does this build-up of autophagosomes disrupt the contractile apparatus in the muscle fibers, it also interferes with enzyme replacement therapy by acting as a sink for the recombinant enzyme and preventing its efficient delivery to the lysosomes. Our data indicate that a re-examination of the presumed pathological mechanism in Pompe disease is necessary, and suggest that successful treatment of patients with Pompe disease will require consideration of the dramatic failure of autophagy that occurs in this disease.
Figures

References
-
- Amalfitano A, Bengur AR, Morse RP, Majure JM, Case LE, Veerling DL, et al. Recombinant human acid α-glucosidase enzyme therapy for infantile glycogen storage disease Type II: results of a Phase I/II clinical trial. Genet Med. 2001;3:132–138. - PubMed
-
- Berg TO, Fengsrud M, Stromhaug PE, Berg T, Seglen PO. Isolation and characterization of rat liver amphisomes. Evidence for fusion of autophagosomes with both early and late endosomes. J Biol Chem. 1998;273:21883–21892. - PubMed
-
- Fukuda T, Ewan L, Bauer M, Mattaliano RJ, Zaal K, Ralston E, et al. Dysfunction of endocytic and autophagic pathways in a lysosomal storage disease. Ann Neurol. 2006b;59:700–708. - PubMed
-
- Fukuda T, Roberts A, Ahearn M, Zaal K, Ralston E, Plotz PH, et al. Autophagy and lysosomes in Pompe disease. Autophagy. 2006c;2:318–320. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous