

Increased superoxide in vivo accelerates age-associated muscle atrophy through mitochondrial dysfunction and neuromuscular junction degeneration
- PMID: 20040516
- PMCID: PMC2987499
- DOI: 10.1096/fj.09-146308
Increased superoxide in vivo accelerates age-associated muscle atrophy through mitochondrial dysfunction and neuromuscular junction degeneration
Abstract
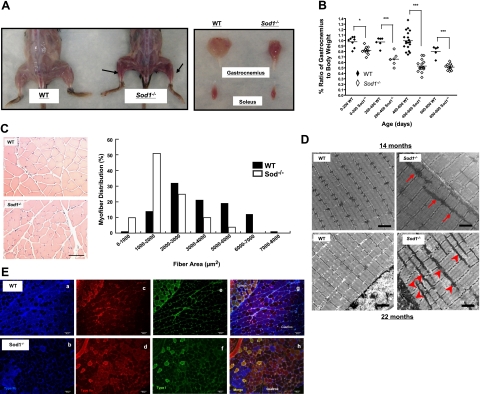
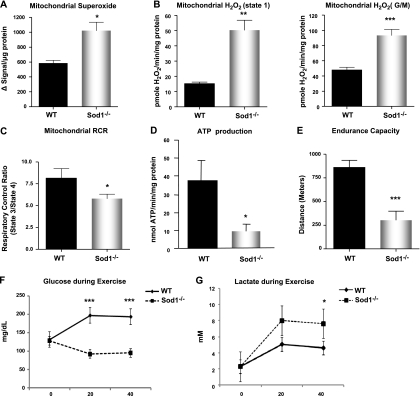
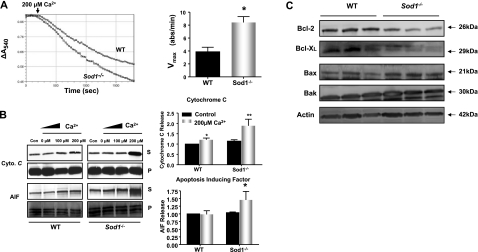
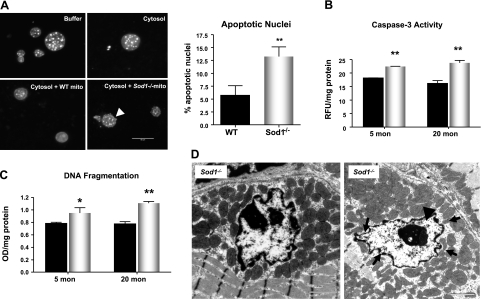
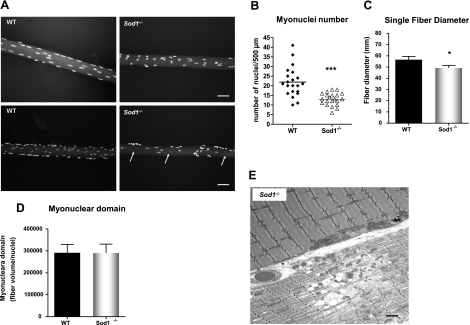
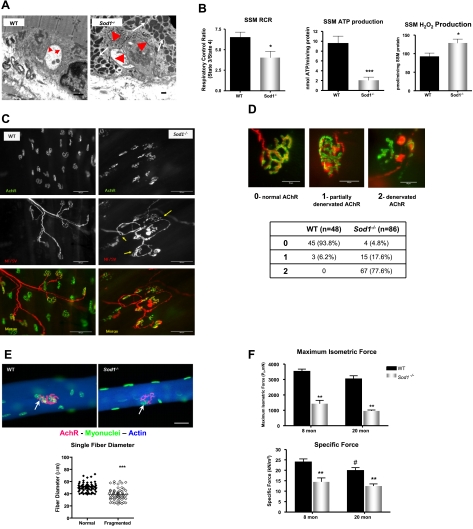
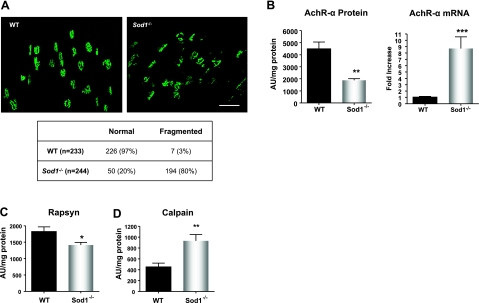
Oxidative stress has been implicated in the etiology of age-related muscle loss (sarcopenia). However, the underlying mechanisms by which oxidative stress contributes to sarcopenia have not been thoroughly investigated. To directly examine the role of chronic oxidative stress in vivo, we used a mouse model that lacks the antioxidant enzyme CuZnSOD (Sod1). Sod1(-/-) mice are characterized by high levels of oxidative damage and an acceleration of sarcopenia. In the present study, we demonstrate that muscle atrophy in Sod1(-/-) mice is accompanied by a progressive decline in mitochondrial bioenergetic function and an elevation of mitochondrial generation of reactive oxygen species. In addition, Sod1(-/-) muscle exhibits a more rapid induction of mitochondrial-mediated apoptosis and loss of myonuclei. Furthermore, aged Sod1(-/-) mice show a striking increase in muscle mitochondrial content near the neuromuscular junctions (NMJs). Despite the increase in content, the function of mitochondria is significantly impaired, with increased denervated NMJs and fragmentation of acetylcholine receptors. As a consequence, contractile force in aged Sod1(-/-) muscles is greatly diminished. Collectively, we show that Sod1(-/-) mice display characteristics of normal aging muscle in an accelerated manner and propose that the superoxide-induced NMJ degeneration and mitochondrial dysfunction are potential mechanisms of sarcopenia.
Figures
References
-
- Mansouri A, Muller F L, Liu Y, Ng R, Faulkner J, Hamilton M, Richardson A, Huang T T, Epstein C J, Van Remmen H. Alterations in mitochondrial function, hydrogen peroxide release and oxidative damage in mouse hind-limb skeletal muscle during aging. Mech Ageing Dev. 2006;127:298–306. - PubMed
-
- Moylan J S, Reid M B. Oxidative stress, chronic disease, and muscle wasting. Muscle Nerve. 2007;35:411–429. - PubMed
-
- Lexell J, Taylor C C, Sjostrom M. What is the cause of the ageing atrophy? Total number, size and proportion of different fiber types studied in whole vastus lateralis muscle from 15- to 83-year-old men. J Neurol Sci. 1988;84:275–294. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
