

Overlapping syndrome with familial partial lipodystrophy, Dunnigan variety and cardiomyopathy due to amino-terminal heterozygous missense lamin A/C mutations
- PMID: 20041886
- PMCID: PMC3150739
- DOI: 10.1111/j.1399-0004.2009.01350.x
Overlapping syndrome with familial partial lipodystrophy, Dunnigan variety and cardiomyopathy due to amino-terminal heterozygous missense lamin A/C mutations
Abstract
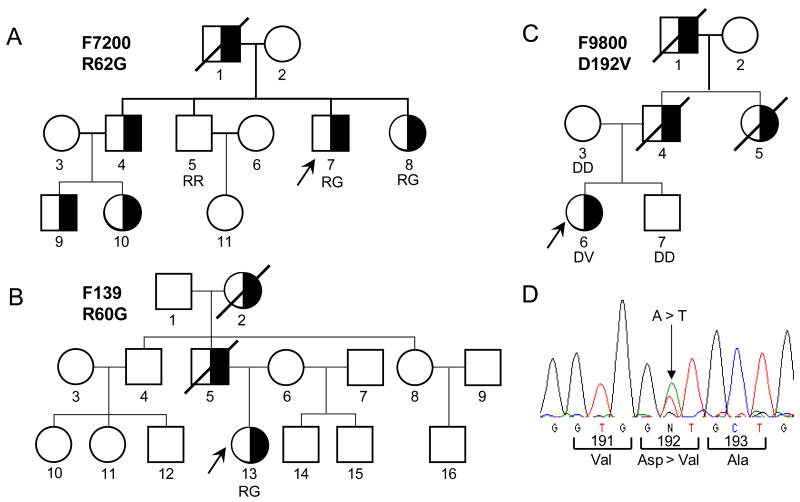
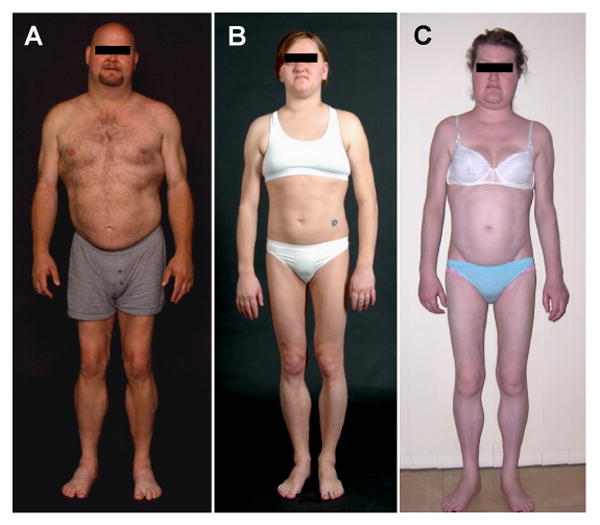
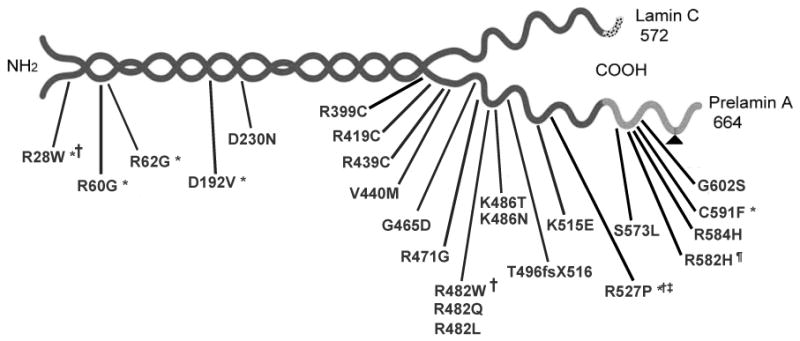
Familial partial lipodystrophy, Dunnigan variety (FPLD) is a well-recognized autosomal dominant disorder due to heterozygous missense mutations in lamin A/C (LMNA) gene. Most of the FPLD patients harbor mutations in the C-terminal of the lamin A/C and do not develop cardiomyopathy. On the other hand, affected subjects from three FPLD pedigrees with heterozygous R28W, R60G and R62G LMNA mutations in the amino-terminal had associated cardiomyopathy presenting as premature onset of congestive heart failure, dilated cardiomyopathy and conduction system disturbances. We report three new FPLD pedigrees presenting with cardiomyopathy associated with heterozygous LMNA mutations in the amino-terminal region. Two of them had previously reported R60G and R62G mutations and one has a novel D192V mutation. Affected subjects belonging to the pedigree with heterozygous R62G mutation had atrial fibrillation and required pacemaker implantation. The affected subjects from the other pedigrees with R60G and D192V mutations developed severe cardiomyopathy requiring defibrillator implantation and cardiac transplantation before 30 years of age in some and premature death in the fourth decade in others. Thus, our report provides further evidence of association of a multisystem dystrophy syndrome in FPLD patients harboring amino-terminal mutations in LMNA. Increased understanding of the genotype-phenotype association might help devise clinical strategies aimed at preventing devastating manifestations of cardiomyopathy including heart failure, arrhythmias and sudden death. Furthermore, the underlying molecular mechanisms by which these amino-terminal mutations cause lipodystrophy as well as cardiomyopathy remain to be understood.
Figures
Similar articles
-
Multisystem dystrophy syndrome due to novel missense mutations in the amino-terminal head and alpha-helical rod domains of the lamin A/C gene.Am J Med. 2002 May;112(7):549-55. doi: 10.1016/s0002-9343(02)01070-7. Am J Med. 2002. PMID: 12015247
-
Mutational and haplotype analyses of families with familial partial lipodystrophy (Dunnigan variety) reveal recurrent missense mutations in the globular C-terminal domain of lamin A/C.Am J Hum Genet. 2000 Apr;66(4):1192-8. doi: 10.1086/302836. Am J Hum Genet. 2000. PMID: 10739751 Free PMC article.
-
Lamin A/C gene: sex-determined expression of mutations in Dunnigan-type familial partial lipodystrophy and absence of coding mutations in congenital and acquired generalized lipoatrophy.Diabetes. 2000 Nov;49(11):1958-62. doi: 10.2337/diabetes.49.11.1958. Diabetes. 2000. PMID: 11078466
-
A-type lamin-linked lipodystrophies.Novartis Found Symp. 2005;264:166-77; discussion 177-82, 227-30. Novartis Found Symp. 2005. PMID: 15773753 Review.
-
Mutations in the LMNA gene encoding lamin A/C.Hum Mutat. 2000 Dec;16(6):451-9. doi: 10.1002/1098-1004(200012)16:6<451::AID-HUMU1>3.0.CO;2-9. Hum Mutat. 2000. PMID: 11102973 Review.
Cited by
-
Looking at New Unexpected Disease Targets in LMNA-Linked Lipodystrophies in the Light of Complex Cardiovascular Phenotypes: Implications for Clinical Practice.Cells. 2020 Mar 20;9(3):765. doi: 10.3390/cells9030765. Cells. 2020. PMID: 32245113 Free PMC article.
-
Phenotypic Differences Among Familial Partial Lipodystrophy Due to LMNA or PPARG Variants.J Endocr Soc. 2022 Oct 11;6(12):bvac155. doi: 10.1210/jendso/bvac155. eCollection 2022 Oct 26. J Endocr Soc. 2022. PMID: 36397776 Free PMC article.
-
Approach to the Patient With Lipodystrophy.J Clin Endocrinol Metab. 2022 May 17;107(6):1714-1726. doi: 10.1210/clinem/dgac079. J Clin Endocrinol Metab. 2022. PMID: 35137140 Free PMC article.
-
Clinical review#: Lipodystrophies: genetic and acquired body fat disorders.J Clin Endocrinol Metab. 2011 Nov;96(11):3313-25. doi: 10.1210/jc.2011-1159. Epub 2011 Aug 24. J Clin Endocrinol Metab. 2011. PMID: 21865368 Free PMC article. Review.
-
Exploring the pathophysiology behind the more common genetic and acquired lipodystrophies.J Hum Genet. 2014 Jan;59(1):16-23. doi: 10.1038/jhg.2013.107. Epub 2013 Oct 24. J Hum Genet. 2014. PMID: 24152769 Review.
References
-
- Jacob KN, Garg A. Laminopathies: multisystem dystrophy syndromes. Molecular genetics and metabolism. 2006;87:289–302. - PubMed
-
- Rankin J, Ellard S. The laminopathies: a clinical review. Clin Genet. 2006;70:261–274. - PubMed
-
- Garg A. Acquired and inherited lipodystrophies. N Engl J Med. 2004;350:1220–1234. - PubMed
-
- Haque WA, Oral EA, Dietz K, et al. Risk factors for diabetes in familial partial lipodystrophy, Dunnigan variety. Diabetes Care. 2003;26:1350–1355. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
